
Resumen
Presentación del caso. Paciente de 44 años de sexo femenino, sin ninguna enfermedad de base preexistente, con una historia de aproximadamente diez meses de presentar lesiones eritemato-descamativas pruriginosas inicialmente localizadas en extremidades inferiores y que luego se generalizaron en todo el cuerpo, asociándose a la pérdida de peso de aproximadamente 15 kg. Intervención terapéutica. El manejo inicial consistió en corticoides tópicos y antihistamínicos orales con poca respuesta clínica. Se inició el estudio por dermatología y se confirmó el diagnóstico inicial de neoplasia cutánea maligna de células T. Luego se realizó el frotis de médula ósea, en el que se identificaron células «cerebriformes» que confirmaron el diagnóstico de síndrome de Sézary. La paciente recibió esquema de quimioterapia ciclofosfamida, doxorrubicina, vincristina, etopósido y prednisona. Evolución clínica. La respuesta inicial fue favorable, con alta hospitalaria y seguimiento en la consulta externa. Transcurridos tres meses de tratamiento, la paciente consultó por episodio febril, tos productiva más distrés respiratorio asociado a estertores basales bilaterales, presentó insuficiencia respiratoria y durante la inducción a la ventilación mecánica sufrió un paro cardiorrespiratorio y falleció.
Respiratory failure in a patient with Sézary syndrome.
Case presentation. A 44-year-old female patient, with no preexisting underlying disease, with a history of approximately ten months of presenting pruritic erythematous-desquamative lesions initially localized in the lower extremities and later generalized throughout the body, associated with weight loss of 15 kg. Treatment. Initial management consisted of topical corticosteroids and oral antihistamines with little clinical response. A dermatology work-up was initiated, and the initial diagnosis of malignant T-cell neoplasm was confirmed. A bone marrow smear was performed, in which "cerebriform" cells were identified, confirming the diagnosis of Sézary syndrome. The patient received cyclophosphamide, doxorubicin, vincristine, etoposide, and prednisone chemotherapy. Outcome. The initial response was favorable, with hospital discharge and outpatient follow-up. After three months of treatment, the patient consulted for a febrile episode, productive cough plus respiratory distress associated with bilateral basal rales, presented respiratory failure, and during induction of mechanical ventilation suffered cardiorespiratory arrest and died.
Introducción
El linfoma cutáneo de células T es un término general para identificar los linfomas no Hodgkin de células T que afectan principalmente a la piel. Hay muchos subtipos de linfoma cutáneo de células T, entre los más comunes son: la micosis fungoide y el síndrome de Sézary (SS), pueden simular trastornos benignos de la piel convirtiéndose en un desafío diagnóstico para los médicos dermatólogos1,2
El SS y la micosis fungoide son entidades íntimamente relacionadas, se considera la micosis fungoide la forma indolente y el SS su fase leucémica agresiva, por lo que existe la hipótesis que el SS puede evolucionar gradualmente a partir de una micosis fungoide u ocurrir de forma espontánea, aunque actualmente algunos autores los consideran entidades diferentes2.
El SS es una neoplasia maligna originada en los linfocitos T, que compromete la piel y puede extenderse a la médula ósea, linfocitos sanguíneos, ganglios linfáticos y diversos órganos, se caracteriza por eritrodermia, adenopatías superficiales y células atípicas en sangre1.
En cuanto a la distribución epidemiológica de este síndrome se sabe que predomina en hombres con una proporción 2:1, en las edades entre los 60 y 70 años1,3, tiene una tasa anual de incidencia de uno por cada diez millones, representando el 3 % de todos los linfomas cutáneos4. Además, afecta en mayor medida a la población blanca en relación a los afroamericanos y no presenta predisposición genética2.
La prevalencia de linfomas cutáneos primarios registrada entre 1986 y 2002 en Holanda y Austria fue del 3 %5, lo que representa datos similares a los reportados por la Organización Mundial de la Salud, Organización para la Investigación y el Tratamiento del Cáncer (WHO/EORTC por sus siglas del inglés)6. En Argentina, se reportó una prevalencia del 0,13 % en un centro dermatológico especializado, en el periodo de 2006 a 20167.
Presentación del caso
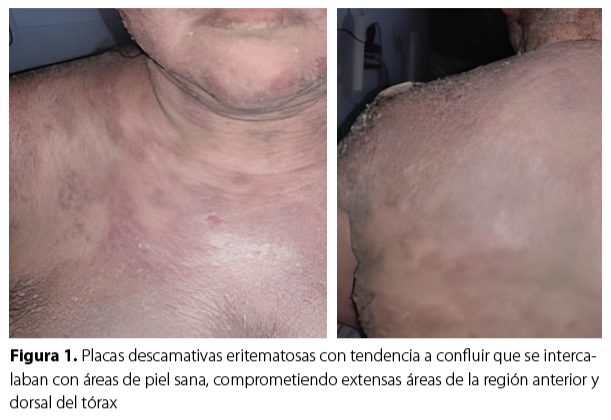
Una paciente femenina de 44 años, sin ninguna enfermedad de base preexistente, sin antecedentes familiares ni quirúrgicos. Consultó con historia de diez meses de prurito generalizado, sin lesiones cutáneas evidentes, acompañado de episodios febriles no cuantificados, fatiga y disminución del apetito, con pérdida de peso aproximadamente de 15 kg, lo que originó múltiples consultas en las que le habían indicado pruebas de laboratorio y había recibido tratamiento con antihistamínicos orales. Además, tenía dos meses de haber iniciado con lesiones cutáneas descamativas, pruriginosas, progresivas y generalizadas (Figura 1 y 2), recibió tratamiento con antihistamínicos orales y esteroides tópicos sin mejoría. En cuanto al examen físico se destaca: xerosis marcada, signos de grataje, eritema generalizado intenso y adenopatías cervicales e inguinales palpables.
Fue evaluada por un dermatólogo de la práctica profesional privada que decidió realizar una biopsia de piel de región dorsal y anterior de tórax. Los resultados de la biopsia de piel con valoración de inmunohistoquímica (IHQ) reveló: CD20 negativo, CD3 positivo, CD5 positivo, CD7 negativo, CD8 positivo en un 10 % con diagnóstico de neoplasia cutánea maligna de células T (Síndrome de Sézary).
Debido a la persistencia del proceso febril y al empeoramiento del estado general de la paciente, se decidió trasladarla al Hospital Médico Quirúrgico del Instituto Salvadoreño del Seguro Social (ISSS), donde las pruebas de laboratorio reportaron marcada leucocitosis (reacción leucemoide), en frotis de sangre periférica: línea eritrocitaria disminuida con anemia moderada, normocítica, normocrómica, células inmaduras del 8 % al 10 % con características de células tipo linfocitos grandes con abundante citoplasma y núcleos irregulares (Tabla 1).
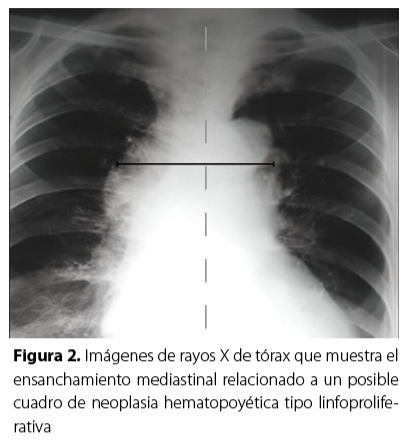
En la radiografía de tórax se describió ensanchamiento mediastinal sin evidencia de lesiones consolidativas en ambas cadenas yugulares, triángulos posteriores del cuello, supraclaviculares e inguinales sin asociarse con áreas de necrosis ni calcificaciones, estos hallazgos generaron la sospecha de un síndrome linfoproliferativo (Figura 3).
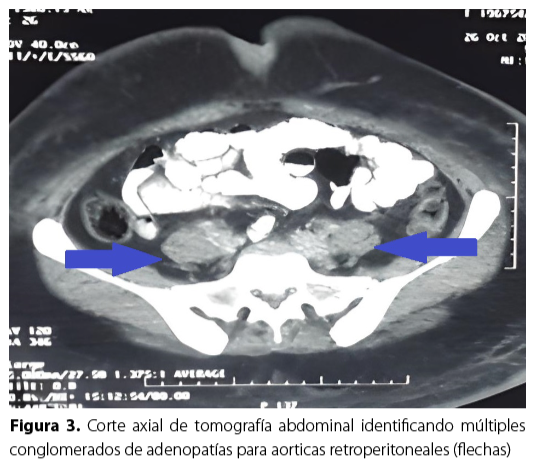
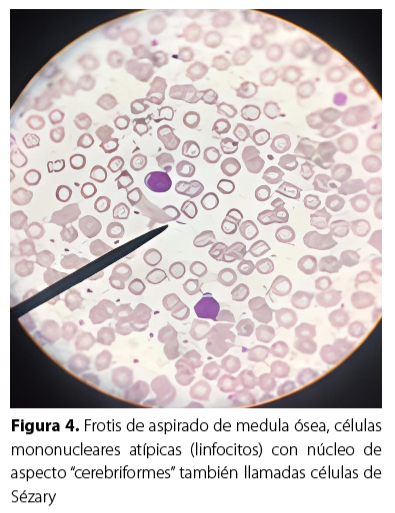
A su ingreso en el servicio hospitalario fue evaluada por la unidad de hematología, donde se le indicó el aspirado de medula ósea para procesamiento de frotis y biopsia (Figura 4), que reportó la presencia de hipercelularidad con presencia de megacariocitos, serie mieloide con 60 % a predominio de formas adultas, aunque con leve aumento de formas juveniles más granulación tóxica; la serie eritroide con forma y tamaño normal pero disminuida en 20 %; serie linfoide con 20 % presentaban núcleos irregulares, algunas con cromatina condensada y aspecto «cerebriformes» hallazgo patognomónico en esta enfermedad. La biopsia de médula ósea fue negativa para linfoma.
La citometría de flujo reportó que el 76 % de todos los eventos son linfocitos, el 97 % de los linfocitos T son CD4 y expresan CD3, CD4, CD5, CD2, CD25 heterogéneo y CD7 negativo, CD34, CD10 y CD56 negativos con lectura por inmunofenotipo de linfoma-leucemia de células T del adulto o linfoma anaplásico.
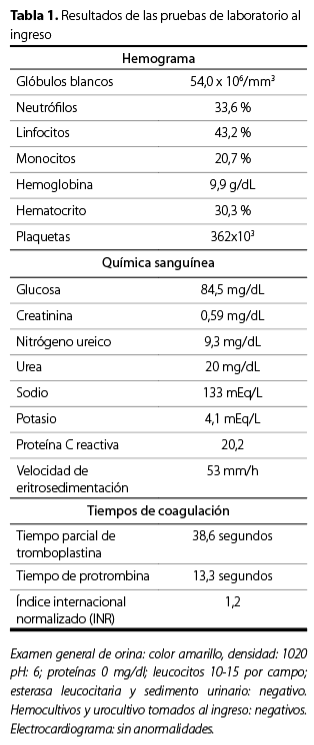
Tabla 1. Resultados de las pruebas de laboratorio al ingreso
|
Hemograma |
|
|
Glóbulos blancos |
54,0 x 106/mm3 |
|
Neutrófilos |
33,6 % |
|
Linfocitos |
43,2 % |
|
Monocitos |
20,7 % |
|
Hemoglobina |
9,9 g/dL |
|
Hematocrito |
30,3 % |
|
Plaquetas |
362×103 |
|
Química sanguínea |
|
|
Glucosa |
84,5 mg/dL |
|
Creatinina |
0,59 mg/dL |
|
Nitrógeno ureico |
9,3 mg/dL |
|
Urea |
20 mg/dL |
|
Sodio |
133 mEq/L |
|
Potasio |
4,1 mEq/L |
|
Proteína C reactiva |
20,2 |
|
Velocidad de |
53 mm/h |
|
Tiempos de coagulación |
|
|
Tiempo parcial de |
38,6 segundos |
|
Tiempo de protrombina |
13,3 segundos |
| Índice internacional |
1,2 |
Examen general de orina: color amarillo, densidad: 1020 pH: 6; proteínas 0 mg/dL; leucocitos 10-15 por campo; esterasa leucocitaria y sedimento urinario: negativo. Hemocultivos y urocultivo tomados al ingreso: negativos. Electrocardiograma: sin anormalidades.
Intervención terapéutica
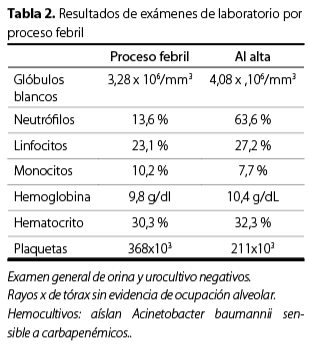
Se indicó el inicio del esquema quimioterapéutico disponible a nivel institucional, con ciclofosfamida, doxorrubicina, vincristina, etopósido y prednisona (CHOEP). Cinco días posteriores al inicio de la quimioterapia, presentó picos febriles de 38,5 °C y se reportó una leucopenia con neutropenia asociada a la quimioterapia, debido a esto, se implementó aislamiento invertido y se inició un esquema antibiótico profiláctico con 2 g de ceftriaxona, vía intravenosa cada día, más 900 mg de clindamicina, vía oral cada ocho horas; los picos febriles persistieron y de forma preliminar se identificó Acinetobacter baumannii, con sensibilidad a carbapenémicos, en los hemocultivos (Tabla 2), este dato se confirmó en el reporte definitivo. Posteriormente, se interconsultó con el servicio de infectología, que indicó el tratamiento por 14 días con 500 mg de imipenem, vía intravenosa, cada seis horas por 14 días. La fiebre cedió al segundo día de tratamiento.
Tabla 2. Resultados de exámenes de laboratorio por proceso febril
|
|
Proceso febril |
Al alta |
|
Glóbulos blancos |
3,28 x 106/mm3 |
4,08 x ,106/mm3 |
|
Neutrófilos |
13,6 % |
63,6 % |
|
Linfocitos |
23,1 % |
27,2 % |
|
Monocitos |
10,2 % |
7,7 % |
|
Hemoglobina |
9,8 g/dl |
10,4 g/dL |
|
Hematocrito |
30,3 % |
32,3 % |
|
Plaquetas |
368×103 |
211×103 |
Examen general de orina y urocultivo negativos.
Rayos x de tórax sin evidencia de ocupación alveolar.
Hemocultivos: aíslan Acinetobacter baumannii sensible a carbapenémicos..
Evolución clínica
La paciente recibió el alta hospitalaria debido a la mejoría clínica con un aumento en el número de glóbulos blancos (Tabla 2). Además, se indicó el plan de seguimiento en consulta externa por la especialidad de hematología.
Transcurrido tres meses de tratamiento oncológico, la paciente consultó por una semana de episodios febriles no cuantificados, tos productiva y disnea. En el examen físico se encontró: estertores basales bilaterales y tiraje subcostal, debido a esto se decidió realizar el manejo de la paciente en el área de máxima urgencia, donde presentó episodios de desaturación y franco distrés respiratorio, por lo que se decidió iniciar con ventilación mecánica, durante la inducción la paciente presentó un paro cardiorrespiratorio que no se revirtió con las maniobras de reanimación y falleció.
Diagnóstico clínico
La presencia de lesiones cutáneas asociadas a las células de Sézary en frotis de aspirado de médula ósea y la expresión aberrante de marcadores en linfocitos T por citometría de flujo confirmaron el diagnóstico de síndrome de Sézary como variante de linfoma cutáneo de células T, finalmente complicada con una insuficiencia respiratoria.
Discusión
El SS fue descrito por primera vez en 1938, por Sézary y Bouvrian, quienes describieron la tríada del síndrome de Sézary caracterizada por: eritrodermia, adenopatías generalizadas y presencia de células T neoplásicas en piel, ganglios linfáticos y sangre periférica6,8.
El SS corresponde a las etapas IVA2 e IVB del linfoma cutáneo de células T (T 1-4, N 0-3, M 0-1, B 0-2) según la estadificación y clasificación específica TNMB propuesta desde 2007 por el consenso entre la Sociedad Internacional de Linfomas Cutáneos y el grupo de trabajo sobre linfoma cutáneo de la Organización Europea para la Investigación y el Tratamiento del Cáncer (ISCL/EORTC)9-11.
En el SS los pacientes presentan eritrodermia extensa e infiltrativa manifestándose a menudo con facies leonina y prurito intenso, pudiendo presentar también alopecia, ectropión, queratodermia palmo plantar leve y onicodistrofia ungueal2,9. Se reportan además adenopatías, hepatoesplenomegalia, asociado a más de 1000/mm3 (o >10 %) células mononucleares atípicas circulantes en el torrente sanguíneo con núcleo de aspecto «cerebriformes» (células de Sézary), estas células pueden ser: CD4+, CD7-, CD26- con una proporción de células T CD4+/CD8+ >107,9.
En su etiopatogenia se han involucrado tanto factores genéticos, en particular, se presentan reordenamientos en la región 6q23-27 que conducen a alteraciones en el proto-oncogén MYB y en el gen de la subunidad alfa-2 del receptor de la interleucina-22 (IL22RA2)4; factores infecciosos como los virus linfotrópico de células T humano tipo 1, infección por virus de Epstein-Barr, citomegalovirus y virus herpes humano tipo 8; factores inmunológicos entre los que se encuentra la falta de regularización de citocinas, las cuales influyen en las células tumorales, donde las células T CD4 (Th2) y sus clonas producen IL-4, IL-5, IL-6 e IL-10; y factores ambientales, sin embargo, su etiología sigue sin aclararse7.
Los criterios de diagnóstico del SS son: eritrodermia en el 80 % de la superficie corporal, recuento anormal de linfocitos, presencia de células de Sézary > 1000 células/mm³ y aumento de células CD4+ en sangre periférica, tener un cociente CD4/CD8 > 10,5 % a 35 %.7,11. La biopsia de la piel puede no ser concluyente en un poco más de la mitad de los casos (60 %)12. También es considerado suficiente para el diagnóstico la evidencia de expansión clonal de CD4+/CD7− ≥ 40 % o CD4+/CD26− ≥El 30 %9.
El diagnóstico diferencial del SS incluye la micosis fungoide, la psoriasis, la pitiriasis rubra pilaris, la dermatitis, el síndrome hipereosinofílico y la leucemia de células T del adulto, trastornos cutáneos primarios como escabiosis, reacciones adversas a medicamentos y enfermedad de injerto contra huésped2,7,13. Es frecuente el retraso en el diagnóstico (hasta seis años de su presentación inicial) ya que puede imitar enfermedades inflamatorias benignas6.
La evaluación inicial de estos pacientes incluye: hemograma, función renal, pruebas de función hepática, lactato deshidrogenasa (LDH), radiografías del tórax, escáner (TAC), RM y PET-TAC, y una biopsia inicial de los ganglios linfáticos6.
El tratamiento depende del estadio de la enfermedad, los pacientes con estadio IA-IIA inician con terapias dirigidas a piel, como esteroides tópicos o fototerapia (psoraleno-UV-A [PUVA] o UVB de banda estrecha). En aquellos con estadio IIB se puede utilizar radioterapia localizada si son lesiones únicas, o gemcitabina o doxorrubicina en monoterapia en lesiones múltiples2,7,9. En pacientes con etapas avanzadas puede considerarse la quimioterapia con doxorrubicina liposomal, gemcitabina o alemtuzumab y en caso de ser jóvenes, se debe considerar el trasplante de progenitores hematopoyéticos o terapia con mogamulizumab un anticuerpo monoclonal dirigido contra el receptor de quimiocinas C-C 4 (CCR4) un receptor de superficie celular transmembrana para las quimiocinas CCL17 y CCL22, que desempeñan un papel en la migración celular y el tráfico de varias subpoblaciones de linfocitos que se dirigen hacia la piel12,14,15.
El pronóstico es malo, se reporta una supervivencia aproximadamente de cinco años no mayores al 30-40 %9,11.
Aspectos éticos
Este informe de caso se realizó conforme a los principios de la Declaración de Helsinki. Se consideró la confidencialidad y no maleficencia de los pacientes.
Agradecimiento
Al Departamento de Hematología del Hospital Médico Quirúrgico del Instituto Salvadoreño del Seguro Social, quienes ayudaron a la preparación e interpretación de los frotis estudiados, así como al departamento de Radiología e Imágenes del Hospital Médico Quirúrgico del Instituto Salvadoreño del Seguro Social por su colaboración en la interpretación de los estudios de imagen presentados.
Financiamiento
Los autores declaran no tener fuentes de financiamiento.
- Entendiendo el linfoma cutáneo de células T. Research foundation. Estados Unidos.2022. Disponible en: https://lymphoma.org/wp-content/uploads/2023/03/Entendiendo_el_linfoma_cutaneo_de_celulas_T.pdf
- Vakiti A, Padala S, Singh D. Sézary Syndrome. PubMed. 2023. https://pubmed.ncbi.nlm.nih.gov/29763049/
- Piña Y, Piña J, Hernández D, Fernández M, Darias C. Síndrome de Sézary. Presentación de un caso. Scielo. 2018;40(2):e 1684-1824. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1684-18242018000200022
- Orphanet. Síndrome de Sézary. Orphanet versión. 2023. Fecha de consulta: 03 de septiembre de 2023. Disponible en: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=3401&Disease_Disease_Search_diseaseGroup=sindrome-de-sezary&Disease_Disease_Search_diseaseType=Pat&Enfermedad(es)/grupo%20de%20enfermedades=S-ndrome-de-Sezary&title=S%EDndrome%20de%20Sezary&search=Disease_Search_Simple
- Rein W, Elaine S, Gunter B, Lorenzo C, Berti E, Steven H. et al Clasificación de la EORTC de la OMS para los linfomas cutáneos. 2005;105(10):3768-3785. DOI: 10.1182/blood-2004-09-3502
- Alonso B, Arias M, Abeldaño A. Síndrome de Sézary: Etiopatogenia, epidemiología, clínica, diagnóstico y pronóstico. Dermatol. Argent. 2020;26(1):2-10. DOI: 10.47196/da.v26i1.2059
- Domínguez M, Ramos J, Vidal A. Síndrome de Sézary, Comunicación de un caso. Medigraphic. 2018; Vol. 27(3):96-102. Disponible en: https://www.medigraphic.com/pdfs/derma/cd-2018/cd183d.pdf
- Gómez S. Pérez N. Micosis fungoide y síndrome de Sézary. Actas dermo- Sifiliográficas. 2001; 92(5):193-206. Disponible en: https://www.actasdermo.org/es-micosis-fungoide-sindrome-sezary-articulo-13014774
- Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, Wood GS, Willemze R, Demierre M-F, Pimpinelli N, et al. Clinical End Points and Response Criteria in Mycosis Fungoides and Sézary Syndrome: A Consensus Statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. JCO. 2011;29(18):2598-2607. DOI: 10.1200/JCO.2010.32.0630
- Pujol RM, Gallardo F. Linfomas cutáneos. Parte I: micosis fungoide, síndrome de Sézary y proliferaciones linfoides cutáneas CD30 positivas. Actas Dermo-Sifiliográficas. 2021;112(1):14-23. DOI: 10.1016/j.ad.2020.08.003
- Falkenhain-López D, Muniesa C, Estrach MT, Morillo-Andújar M, Peñate Y, Acebo E, Pujol RM, García-Muret MP, Machan S, Medina S, et al. Registro de linfomas cutáneos primarios (RELCP) de la AEDV: datos tras 5 años de funcionamiento. Actas Dermo-Sifiliográficas. 2023;114(4):291-298. DOI: 10.1016/j.ad.2022.11.010
- Acosta A, Alperovich M, Miranda A, Arias M, Bertoncini C, Car-mona L. et al. Consenso Linfomas Cutáneos Primarios. Argentina: Sociedad Argentina de Dermatología; 2018. 34p. Disponible en: https://docplayer.es/107537347-Consenso-publicacion-de-la-sociedad-argentina-de-dermatologia-linfomas-cutaneos-primarios-actualizacion-2018.html
- Larocca C, Kupper T. Mycosis Fungoides and Sézary Syndrome. Hematology/Oncology Clinics of North America. 2019;33(1):103-120. DOI: 10.1016/j.hoc.2018.09.001
- Foss F, Girardi M. Mycosis Fungoides and Sezary Syndrome. Hematology/Oncology Clinics of North America. 2017;31(2):297-315. DOI: 10.1016/j.hoc.2016.11.008
- Nicolay JP, Albrecht JD, Alberti-Violetti S, Berti E. CCR4 in cutaneous T-cell lymphoma: Therapeutic targeting of a pathogenic driver. Eur. J. Immunol. 2021;51(7):1660-1671. DOI: 10.1002/eji.202049043
Citación recomendada: Granados Flores NA, Castillo Hernández GJ, Trejo Ayala RA. Insuficiencia respiratoria en una paciente con síndrome de Sézary. Alerta. 2023;6(2):93-98. DOI: 10.5377/alerta.v6i2.16211







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}