Resumen
La esclerosis múltiple es una enfermedad inflamatoria desmielizante, autoinmune y crónica del sistema nervioso central, caracterizada por la desmielinización y la posterior neurodegeneración secundaria al daño neuronal por pérdida axonal. Actualmente, sigue siendo una enfermedad de etiología desconocida que afecta a más de 2 000 000 personas. Se asocia a diversos factores genéticos y ambientales que aumentan su susceptibilidad y ocurre principalmente en el grupo de edad de 20 a 40 años. Para la elaboración de este artículo, se efectuó una revisión de la bibliografía disponible en bases de datos como PubMed y SagePub. Se seleccionaron artículos originales, revisiones bibliográficas, revisiones sistemáticas y metaanálisis en inglés y español, con el objetivo de realizar una reseña de la esclerosis múltiple, sus antecedentes, epidemiología, manifestaciones clínicas, clasificación, criterios diagnósticos y terapia disponible. Los avances en el tratamiento han logrado mejorar la calidad de vida al reducir la frecuencia y severidad de los brotes, pero la etiología de la enfermedad sigue siendo incierta y sus efectos neurodegenerativos de mal pronóstico.
Multiple sclerosis is a chronic autoimmune demyelinating inflammatory disease of the central nervous system, characterized by demyelination and subsequent neurodegeneration secondary to neuronal damage due to axonal loss. Currently, it is still a disease of unknown etiology that affects more than 2 000 000 people, is associated with various genetic and environmental factors that increase susceptibility, and occurs mainly in the 20-40 age group. For the preparation of this article, a review of the literature available in databases such as PubMed and SagePub. Original articles, bibliographic reviews, systematic reviews, and meta-analyses in English and Spanish were selected in order to review multiple sclerosis, its background, epidemiology, clinical manifestations, classification, diagnostic criteria and available therapy. Advances in treatment have improved the quality of life by reducing the frequency and severity of flares. However, the etiology of the disease remains uncertain, and its neurodegenerative effects are poorly prognosed.
Introducción
La esclerosis múltiple (EM) es una enfermedad inflamatoria desmielinizante autoinmune crónica del sistema nervioso central (SNC), caracterizada por la desmielinización y la posterior neurodegeneración secundaria al daño neuronal por pérdida axonali. La etiología sigue siendo desconocida y se ha asociado a factores que aumentan la susceptibilidad. Su patogénesis está mediada principalmente por linfocitos T (LT ) y linfocitos B (LB), que inician el ciclo de inflamación y desmielinización, seguido de remielinización parcial, neurodegeneración y gliosis en la sustancia blanca del SNCii.
El diagnóstico se basa en una evaluación clínica inicial apoyada por estudios de laboratorio y neuroimagen. Su terapia es multidisciplinaria. Los fármacos utilizados pueden dividirse en función de su objetivo en el curso de la enfermedadii. La EM se presenta mayoritariamente entre los 20 y 40 años, y es más frecuente en mujeresiii,iv. Para esta revisión, se realizó una búsqueda en bases de datos como PubMed y SagePub. Se seleccionaron artículos originales, revisiones bibliográficas, revisiones sistemáticas y metaanálisis en inglés y español, con el objetivo de describir la EM, presentar nuevas orientaciones sobre su patogénesis y establecer el contexto actual en términos de epidemiología, características clínicas, clasificación, criterios diagnósticos y opciones terapéuticas disponibles, que pueden ser relevantes para el futuro entendimiento de esta enfermedad.Discusión
Antecedentes históricos y contexto actual
Las características clínicas de la EM fueron descritas por primera vez por Jean-Martin Charcot, quien distinguió entre el temblor de la parálisis agitada (más tarde denominada enfermedad de Parkinson) y el de la EM. Los tres indicadores más fiables de la EM (temblor intencional, nistagmo y habla escandida) pasaron a conocerse como la tríada de Charcoti. Paralelamente, han surgido nuevos tratamientos, incluyendo terapias modificadoras de la enfermedad que han demostrado eficacia en la reducción de recaídas y la progresión de la discapacidad. A pesar de estos avances, la EM continúa siendo una enfermedad sin cura, con un curso clínico impredecible que varía entre diferentes manifestaciones, lo que subraya la necesidad de seguir investigando para mejorar el manejo y la calidad de vida de los pacientes.
Etiología
El origen de la EM es multifactorial, se relaciona con factores genéticos y ambientales. Entre los que se encuentran vivir en latitudes alejadas del ecuador, deficiencia de vitamina D, exposición a rayos ultravioleta B, infección por herpes virus (EpsteinBarr, herpes simple y varicelazoster), neumonía por micoplasma, obesidad, dieta y tabaquismoiiiv. El riesgo de desarrollar EM en familiares está relacionado con la susceptibilidad de los rasgos genéticos. La proporción de riesgo genético en gemelos monocigóticos con 100% de similitud genética es 25%. En casos de similitud del 50%, como gemelos dicigóticos y familiares de primer grado, el riesgo disminuye al 25%. Asimismo, en familiares de segundo grado, con un 25% de similitud genética, el riesgo es del 12%, mientras que en familiares de tercer grado, con un 12,5% de similitud genética, es inferior al 1%v. En la región del antígeno leucocitario humano (HLA, por sus siglas en inglés) del cromosoma 6, se ha identificado un grupo de genes asociados a un mayor riesgo de EM. Los principales genes implicados residen en el gen HLA-DRB1*15; sin embargo, actualmente, se han identificado más de 200 asociaciones genéticas independientesvi,vii.
Microorganismos, como virus y bacterias, pueden tener antígenos estructuralmente homólogos con componentes de la mielina, como la proteína proteolipídica, la proteína básica de la mielina y/o la glicoproteína asociada a la mielina. Así que, cuando estos patógenos activan las células inmunitarias, podrían desarrollarse lesiones de mielina paralelasviii. Otras hipótesis postulan que la deficiencia de vitaminas, especialmente D y B12, constituye un factor de riesgo.
Recientemente se ha demostrado que la obesidad puede estar relacionada con EM mediante tres teorías. La primera es la teoría inflamatoria, donde se postula que la hipertrofia implicada y posterior hiperplasia de adipocitos en la obesidad se caracteriza por inflamación de bajo grado, en la que se producen altos niveles de mediadores proinflamatorios. La segunda es la teoría hormonal, en la que el exceso de tejido adiposo se acompaña de secreción alterada de adipocinasix. La tercera se relaciona con la deficiencia de vitaminas; la obesidad también conduce a menor disponibilidad de vitamina D, lo cual se correlaciona con un estado proinflamatoriox. Otra posible etiología es la asociación entre cambios en el microbioma intestinal y alteraciones del SNC.
La selectividad de la barrera hematoencefálica (BHE) está relacionada con una microbiota intestinal favorable que puede activar la microglía. En la EM, hay una abundancia de ciertas especies bacterianas como Archaea, al mismo tiempo que disminución o ausencia de otros tipos como los filos Firmicutes y Bacteroidetes. Las asociaciones de ciertos perfiles de microbiota se han relacionado con mayor riesgo de recaídasxi. Los factores dietéticos y de estilo de vida pueden participar en síntomas de la EM modulando el estado inflamatorioxii. Componentes como ácidos grasos, polifenoles y dietas altas en carbohidratos o grasas pueden desencadenar una serie de reacciones inflamatorias.
La persistencia de esta dieta promueve el metabolismo de las células hacia vías biosintéticas, incluyendo la producción de moléculas proinflamatorias como el factor de necrosis tumoral, interleucinas, metaloproteinasas de matriz, prostaglandinas y leucotrienos, que contribuyen a la inflamación sistémica y estrés oxidativoxii. Por el contrario, el ejercicio y las dietas bajas en calorías basadas en verduras, frutas, legumbres, pescado, prebióticos y probióticos actúan sobre receptores nucleares y enzimas que regulan al alza el metabolismo oxidativo y a la baja la síntesis de moléculas proinflamatoriasxiii. Por último, numerosas pruebas sugieren que el tabaquismo, debido a la producción de óxido nítrico y monóxido de carbono podría desempeñar un papel importante en la etiología de la EMxiv-xvi.
Fisiopatología
Esta enfermedad autoinmune está mediada principalmente por LT activados; sin embargo, pruebas recientes han demostrado contribución de LB a la patogénesis. La respuesta inmunológica se inicia por activación periférica de LT autorreactivos tras disminución de la autotolerancia a mielina o a diferentes antígenos del SNC. El desencadenante puede ser un antígeno ambiental (virus), una reacción cruzada entre proteínas endógenas (proteína básica de la mielina) o una proteína exógena patógena (antígeno viral); por mimetismo molecular. Tras la activación de LT reactivos a mielina, estos atraviesan la BHE facilitando la expresión y regulación positiva de moléculas de adhesión, quimiocinas y metaloproteínas de matriz.
Al entrar en SNC, los LT autorreactivos pueden ser reactivados por células presentadoras de antígeno, desencadenando una cascada inflamatoria por la liberación local de citocinas y quimiocinas, el reclutamiento de células inflamatorias adicionales como monocitos, LB y LT, y la activación persistente de macrófagos. Esto resulta en daño de la mielina y pérdida de oligodendrocitos ocasionada por la respuesta inflamatoria en contra de la mielina, así como pérdida axonal que puede producirse durante las primeras fases inflamatorias o cuando los mecanismos de reparación se agotan por la activación persistente de macrófagos y complemento y por efectos indirectos de citocinas proinflamatorias como el factor de necrosis tumoral alfa, el óxido nítrico y las metaloproteinasas de matrizvii. Las características patológicas de la EM son inflamación, desmielinización seguida de remielinización o neurodegeneración y gliosis, que se producen de forma focal o difusa en la sustancia blanca y gris del SNC. Estas características están presentes en la mayoría de los subtipos de EM, aunque varían con el tiempo y entre individuosxvii.
Epidemiología
La prevalencia de la EM es variable, es más alta en Norteamérica y Europa, con 140 y 108 casos por 100 000 habitantes, y la más baja en el África subsahariana y Asia oriental, con 2,1 y 2,2 por 100 000, respectivamentevii. Se estima que la EM afecta a más de 2000 000 de personas en todo el mundo, tiene una predilección femenina a razón de 3:1, se presenta con mayor frecuencia en personas de 20 a 40 años y es la causa no traumática más común de discapacidad neurológica en adultos jóvenesii,xviii,ixx.
Su prevalencia varía según la geografía y la etnia, y aumenta con la latitudxx,xxi. Su incidencia parece haber aumentado en los últimos 100 años, principalmente en mujeres y poblaciones consideradas tradicionalmente de bajo riesgo, como hispanos, asiáticos y afroamericanos. A pesar de las dificultades en la vigilancia, la incidencia de EM es muy baja en africanos y poblaciones nativas de Latinoamérica y Oceaníaxxii,xxiii.
Los estudios sobre migración apoyan el hecho de que la EM puede ser el resultado del contacto con factores de riesgo ambientales en etapas tempranas de la vida (antes de los 15 años). Se ha demostrado que los pacientes adultos inmigrantes a Europa procedentes de países de bajo riesgo, tienen un riesgo bajo de desarrollar EM, mientras que los hijos de inmigrantes a Europa tienen un riesgo mayor de desarrollar la enfermedadxxiv.
Manifestaciones clínicas
Los síntomas y signos clínicos son variables en función de la localización dañada, pudiendo afectar a vías sensitivas, motoras, visuales y tronco encefálicas. Las zonas de lesión más frecuentes son las periventriculares, el nervio óptico, el tronco encefálico, los pedúnculos cerebelosos y la médula espinalxxv. Al inicio, la mayoría de los pacientes debutan con episodios remitentes recurrentes de síntomas y disfunción neurológica que duran más de 24 horas, definidos como reagudizacionesxxvi.
Signos y síntomas en las fases iniciales
Síndrome clínicamente aislado
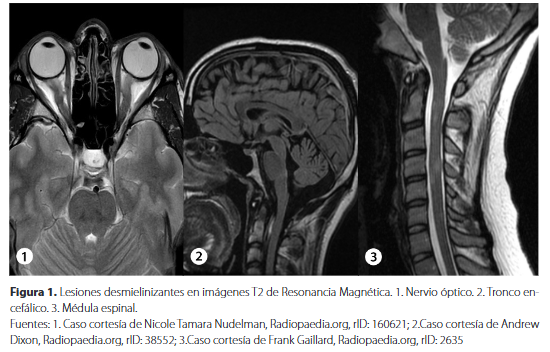
Considerado como el primer evento clínico desmielinizante compatible con EM. Suele ocurrir en adultos jóvenes y afecta a los nervios ópticos, el tronco encefálico o la médula espinal (Figura 1). Aunque los pacientes suelen recuperarse del episodio de presentación, el curso de la EM tras un síndrome clínicamente aislado es variable. Después de 1520 años, un tercio de los pacientes tienen un curso benigno con discapacidad mínima o nula y la mitad desarrollará EM secundaria progresiva con discapacidad crecientexxvii,xxviii.
Neuritis óptica
Es la primera manifestación clínica en el 20% de los pacientes con EM, se produce una pérdida unilateral progresiva de la visión en los primeros días. Puede haber dolor retroocular que empeora con los movimientos oculares, además de disminución de la agudeza visual y alteraciones en la percepción de los colores, especialmente el rojo. Estos síntomas pueden acompañarse de defecto pupilar aferente relativo y escotoma centralixxx. El disco óptico puede aparecer normal (neuritis retrobulbar) o inflamado de forma aguda, puede volverse pálido y atrófico, tiempo después del ataque. Tras un episodio de neuritis óptica unilateral, son frecuentes las recaídas ipsilaterales o contralateralesxxx.
Mielitis aguda
Suele ser parcial y se manifiesta de manera subaguda. Las lesiones se caracterizan por ser pequeñas, suelen estar localizadas en la periferia de la médula espinal y no se extienden más de dos segmentos vertebrales contiguos longitudinalmente. Además, estas lesiones son de localización cervical o dorsal en la mayoría de los casosxxxi. Los síntomas agudos parciales aparecen en horas o días, y consisten en alteraciones sensitivas y motoras que podrían asociarse a una afectación esfinteriana.El daño medular a nivel cervical puede provocar el signo de L’hermitte, el cual es una breve sensación similar a la producida por una descarga eléctrica provocada por la flexión cervicalxxxii. La aparición de una sensación de tensión en forma de banda alrededor del tórax o el abdomen (el cual es conocido como el «abrazo» de la EM), es un síntoma característico de la mielitis que puede sugerir la afectación de las columnas posteriores de la médula espinalixxx.
Síndromes del tronco encefálico
Los síndromes del tronco encefálico pueden manifestarse con síntomas como diplopía, oscilopsia, pérdida de sensibilidad facial, vértigo y disartria. Entre los signos característicos se encuentran la parálisis aislada del sexto par craneal, nistagmo inducido por la mirada o la oftalmoplejía internuclear. La oftalmoplejía internuclear bilateral se considera un signo clínico patognomónico de EMixxx.
Signos y síntomas en las fases establecidas de la enfermedad
Espasticidad
La prevalencia acumulada en pacientes con EM es de aproximadamente 47,5%xxxiii. Los síntomas más comunes asociados a la espasticidad de la EM son la rigidez muscular, los espasmos y las restricciones de la movilidad, que pueden incluir fatiga, dolor y disfunción vesicalxxxi.
Trastornos sensoriales
Los síntomas como el entumecimiento y las parestesias son frecuentes en la mayoría de los pacientes durante el curso de la EM. Estos síntomas pueden sugerir una lesión desmielinizante aguda cuando su duración persiste durante horas o días. El dolor es un síntoma presente en hasta el 54% de los pacientes con EM establecida o diagnosticada recientemente (en los últimos 12 meses). El dolor en la EM puede ser tanto nociceptivo como neuropático. El dolor neuropático puede ser central o periférico y puede estar causado por lesiones cerebrales o de la médula espinalxxxiv.
Subtipos de EM
Esclerosis múltiple recurrente-remitente
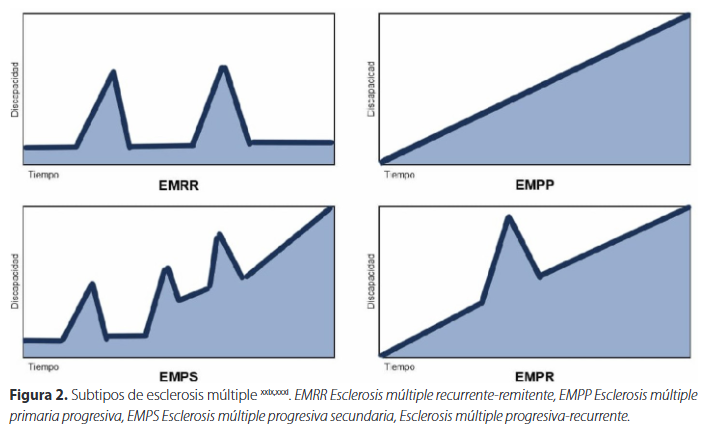
Es el curso clínico más común de la enfermedad, presentándose en aproximadamente el 85% de los casos. Se caracteriza por una alternancia de periodos de disfunción neurológica, conocidos como recaídas, seguidas de una recuperación parcial o completa, y periodos de relativa estabilidad clínica sin nuevos síntomas neurológicos, denominados remisionesixxx,xxxi,xxxv.
La frecuencia de las recaídas difiere en cada caso, pero no suele superar 1,5 recaídas al año. Durante la recaída pueden presentarse diversos síntomas neurológicos, como debilidad, alteración en la sensibilidad, alteración del equilibrio, alteración en la agudeza visual y de la percepción de colores, o visión doble, que duran al menos 24 horas en ausencia de infección o alteración metabólicaxxxvi.
Este fenotipo de la enfermedad debe cumplir un criterio diagnóstico de diseminación en tiempo y espacio, es decir, la presencia de múltiples eventos clínicamente distintos que afectan diferentes partes del SNC separadas por al menos un mesxxxvii. (Figura 2)
Esclerosis múltiple primaria progresiva
Entre el 10 y 20% de los pacientes con EM presentan este fenotipo, caracterizado por una progresión constante de los síntomas neurológicos sin remisiones evidentes desde su inicioxxxi. La progresión no es uniforme a lo largo del curso y es posible que se produzcan recaídas superpuestas, así como periodos de relativa estabilidad. La aparente ausencia de una fase de recaída-remisión en pacientes con esclerosis múltiple primaria progresiva podría atribuirse posiblemente a lesiones individuales que se localizan en regiones clínicamente silentes, que se agregan para acabar induciendo discapacidadxxxvi,xxxviii. (Figura 2).
Esclerosis múltiple progresiva secundaria
Este subtipo inicia como EM recurrente remitente y, tras 10 a 15 años de enfermedad un 50% de pacientes no tratados presentan un deterioro clínico progresivoxxxix. De los factores predictivos de conversión a la forma secundaria progresiva, la edad de inicio es el más importante (a mayor edad, menor tiempo de progresión). El sexo femenino se ha asociado a un mayor tiempo de progresión. Con respecto a los síntomas iniciales, varios estudios han informado de que los síntomas visuales o sensoriales, y ocasionalmente los síntomas relacionados con el tronco encefálico, se asocian con un mayor tiempo hasta la progresión secundaria, mientras que los síntomas relacionados con la médula espinal se asocian con un menor tiempo hasta la progresiónxl. (Figura 2)
Esclerosis múltiple progresiva-recurrente
Se consideraba un subtipo de la EM primaria progresiva que podía presentar recaídas poco frecuentes superpuestas a una progresión lenta. En 2013, la National Multiple Sclerosis Society eliminó la clasificación clínica de esclerosis múltiple progresiva-recurrente. Las personas previamente diagnosticadas se reclasificaron como EM primaria progresiva activas, en la presencia de las recaídas o nuevas lesiones en las imágenes por resonancia magnética (IRM), o no activasxxxv,xxxvi. (Figura 2)
Diagnóstico
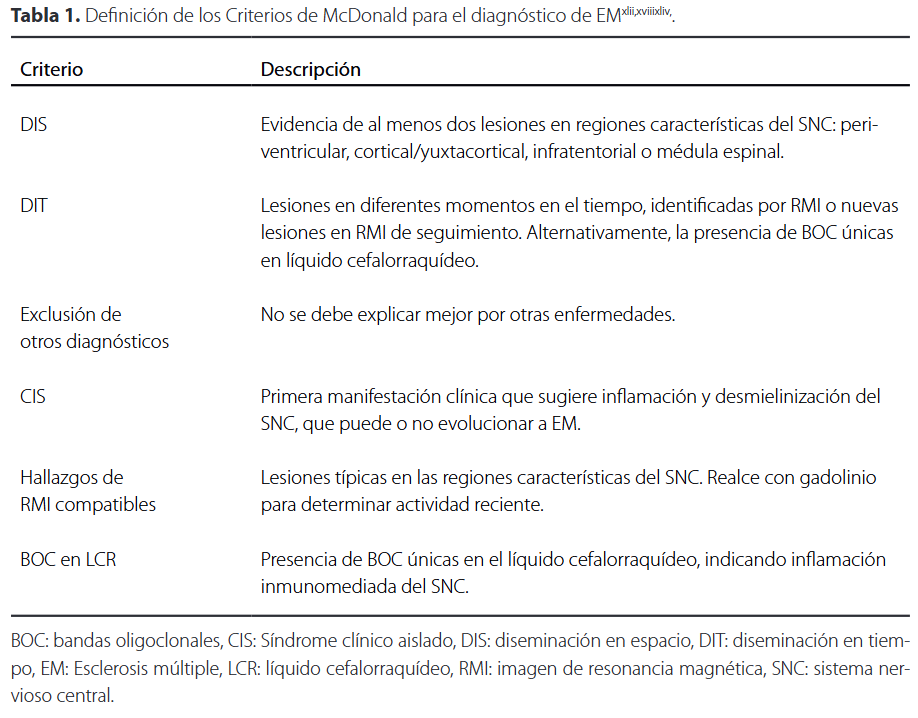
El diagnóstico se basa en la presentación clínica, los hallazgos de neuroimagen, el análisis del líquido cefalorraquídeo (LCR) y los estudios de potenciales evocadosxli. Actualmente, los criterios de McDonald son el estándar diagnóstico. Estos combinan datos clínicos, IRM, y análisis del LCR para establecer un diagnóstico con mayor precisión. Los criterios han sido revisados en varias ocasiones desde su introducción en 2001, con la actualización más reciente en 2017 por la InternationalPanel on Diagnosis of Multiple Sclerosisxlii,xliii. Estos criterios proponen que, para el diagnóstico, es necesario demostrar dos principios fundamentales: la diseminación en el espacio (DIS) y en tiempo (DIT ), descartando cualquier otra causa diagnóstica más probable.
La DIS se refiere a la evidencia de que las lesiones desmielinizantes están presentes en diferentes áreas al detectar lesiones por IRM en al menos dos de las cuatro regiones clásicas: periventricular, cortical o yuxtacortical, infratentorial y medular. La DIT, por su parte, se establece al demostrar que las lesiones ocurrieron en distintos momentos, lo cual puede evidenciarse por la presencia simultánea de lesiones que captan gadolinio y otras que no, o por la aparición de nuevas lesiones en estudios de seguimiento por IRMxliii,xliv. Si se cumplen los criterios postulados (Tabla 1) y no hay una explicación mejor de la presentación clínica, el diagnóstico es EM. Sin embargo, si se sospecha la enfermedad, pero no se cumplen totalmente los criterios, se clasifica como posible EM. Y si durante la evaluación se plantea otro diagnóstico que explique mejor la presentación clínica, el diagnóstico no es EMxliii.
Evaluación de la EM: Escala ampliada del estado de discapacidad (EDSS)
Con el fin de evaluar objetivamente el estado de discapacidad física y cognitiva de los pacientes, se han diseñado pruebas que representan el impacto del curso clínico de la enfermedad. En 1954, Kurtzke desarrolló la primera escala para evaluar la discapacidad física en pacientes con EM, denominada Escala de Estado de Discapacidad, compuesta por ocho sistemas funcionales, que incluyen piramidal, cerebeloso, tronco cerebral, sensorial, vejiga-intestino, visual, cerebral o mental, y otros. Cada sistema funcional tiene una ponderación que oscila entre «0» (hallazgos normales en el examen neurológico) y «5» (hallazgos claros de lesión neurológica); posteriormente, la puntuación máxima se sustituyó por «6».En la actualidad, la Escala Ampliada del Estado de Discapacidad se basa en los resultados de la entrevista clínica y la exploración neurológica. Consta de 20 pasos, inicia en «0» (estado neurológico normal) e incrementa según el deterioro neurológico hasta «10» (muerte por EM)xlv.
Pronóstico
No existen factores pronósticos establecidos para la EM; sin embargo, se han asociado variables de riesgo para la gravedad de la enfermedad. Entre ellos se encuentran algunos componentes demográficos, como ser hombre, tener más de 40 años, ser de origen africano o latinoamericano.Los factores clínicos incluyen la severidad de la recaída, definida como: el aumento de un punto o más en la puntuación Escala Ampliada del Estado de Discapacidad, un incremento de dos puntos o más en un área del sistema funcional, o el incremento de un punto o más en dos áreas del sistema funcional; el uso de esteroides; la hospitalización; ataque multifocal con recuperación parcial e incompleta; y los efectos motores, cerebelosos, esfinterianos y cognitivos del ataque. Además, la frecuencia de recaídas en 25 años, los intervalos cortos entre ataques, y curso de la enfermedad con discapacidad rápidamente progresiva. En el diagnóstico por imagen, inicio con: alta carga de lesiones T2, más lesiones en realce con gadolinio, lesiones T1, lesiones infratentoriales (Figura 3). En el seguimiento: nuevas lesiones T2, una o más lesiones en realce con gadolinioxlvi.
Terapéutica utilizada en el tratamiento de la EM
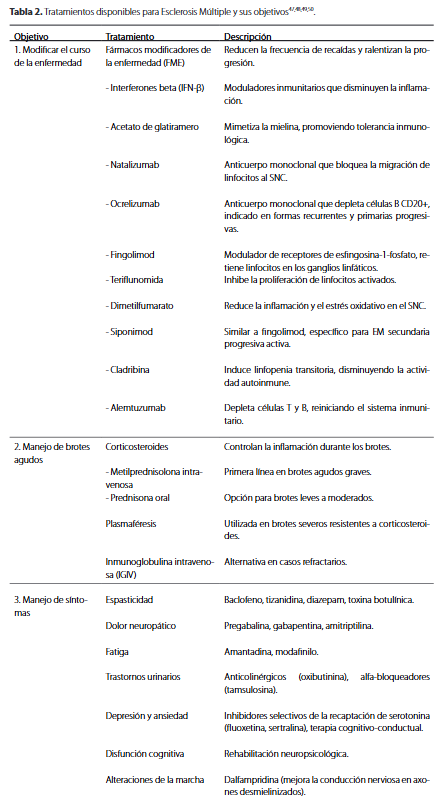
El tratamiento eficaz de la EM requiere un enfoque multidisciplinario para controlar los ataques agudos, manejar el empeoramiento progresivo y tratar los síntomas asociadosxl,xlvii. Los fármacos utilizados pueden dividirse en función de su objetivo en el curso de la enfermedad, y los tratamientos para la EM pueden clasificarse en tres categorías: tratamiento de las recaídas agudas, tratamientos modificadores de la enfermedad (TME) y tratamientos sintomáticos (Tabla 2). Las exacerbaciones agudas son episodios de alteraciones neurológicas focales que duran al menos 24 horas, precedidos de periodos de estabilidad clínica de al menos 30 díasxxvi.
Estas son el resultado de nuevas lesiones en la sustancia blanca; se ha demostrado mediante IRM la unión de gadolinio a lesiones de la sustancia blanca en pacientes con recaídas. El objetivo del tratamiento en pacientes con reagudizaciones de EM es disminuir la duración e intensidad de la disfunción neurológica. Para lograrlo, utilizan principalmente los corticoesteroides (metilprednisolona) y algunas técnicas alternativas como la plasmaféresisxlix. En cuanto al tratamiento a largo plazo, los TME como los interferones beta, acetato de glatiramero, modificadores de la esfingosina-1-fosfato y anticuerpos monoclonales como el natalizumab, modifican el curso de la EM debido a la supresión o modulación de diversos efectos de la función inmune, ejercen actividad antiinflamatoria principalmente en las fases de recaída. Los TME reducen la tasa de recaídas y disminuyen la acumulación de lesiones observadas en la IRM. Además, estabilizan, retrasan y en algunos casos, mejoran modestamente la discapacidad a largo plazo. Sin embargo, estos no son curativosxl,l.
Conclusiones
La EM es una enfermedad compleja y multifactorial que afecta a la población mundial, con patrones geográficos, étnicos y demográficos distintivos. Presenta una mayor prevalencia en mujeres jóvenes de regiones alejadas del ecuador. Desde su descripción inicial, los avances en el conocimiento de su fisiopatología, diagnóstico y manejo, han logrado desarrollar terapias que mejoran la calidad de vida al reducir la frecuencia y severidad de los brotes. Sin embargo, la etiología de la enfermedad sigue siendo incierta, y sus efectos neurodegenerativos de mal pronóstico. El futuro del manejo de la EM se dirige hacia una medicina de precisión que integre datos genómicos, proteómicos y del microbioma, permitiendo un diagnóstico temprano y tratamientos personalizados.
Las investigaciones sobre los loci genéticos asociados, como el HLA-DRB1*15, y su interacción con factores ambientales podrían revolucionar la comprensión de la enfermedad y la predicción del riesgo. Además, la modulación de la microbiota intestinal y los factores dietéticos ofrecen nuevas posibilidades terapéuticas al influir en la regulación inmunológica, como el HLA-DRB1*15, y su interacción con factores ambientales podrían revolucionar la comprensión de la enfermedad y la predicción del riesgo. Además, la modulación de la microbiota intestinal y los factores dietéticos ofrecen nuevas posibilidades terapéuticas al influir en la regulación inmunológica.
En el ámbito terapéutico, los avances en inmunomodulación, dirigidos específicamente a células T y B, junto con el uso de biomarcadores para evaluar la actividad de la enfermedad y la respuesta a tratamientos, permitirán un manejo más efectivo. Además, las estrategias de regeneración y neuroprotección, como la remielinización mediante células madre y moduladores biológicos, presentan un potencial prometedor para abordar la neurodegeneración progresiva. Finalmente, la nanotecnología para la administración dirigida de medicamentos y la identificación de biomarcadores específicos podría transformar el tratamiento actual.
Agradecimiento
A Julio Sotelo y Graciela Ordoñez del departamento de neuroinmunología del Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez, por su asesoría y revisión.
Financiamiento
Esta revisión se realizó con auto financiamiento de los autores.
- Orrell RW. Multiple Sclerosis: The History of a Disease. Journal of the Royal Society of Medicine. 2005;98(6):289-289. DOI:10.1177/014107680509800616
- Dobson R, Giovannoni G. Multiple sclerosis a review. European Journal of Neurology. 2019;26:27-40. DOI:10.1111/ene.13819.
- Alfredsson L, Olsson T. Lifestyle and Environmental Factors in Multiple Sclerosis. Cold Spring Harbor perspectives in medicine. 2019;9(4):a028944. DOI:10.1101/cshperspect.a028944
- Sotelo J, Martínez-Palomo A, Ordoñez G, Pineda B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Annals of neurology. 2008;63(3):303-311. DOI:10.1002/ana.21316
- Cree BA. Multiple sclerosis genetics. Handbook of clinical neurology. 2014;122:193–209. DOI:10.1016/B978-0-444-52001-2.00009-1
- Baranzini SE, Oksenberg JR. The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years. Trends in genetics. 2017;33(12):960–970. DOI:10.1016/j.tig.2017.09.004
- Yamout BI, Alroughani R. Multiple Sclerosis. Seminars in neurology. 392 2018;38(2):212-225. DOI:10.1055/s-0038-1649502
- Liu R, Du S, Zhao L, et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Frontiers in immunology. 2022;13:996469. DOI:10.3389/fimmu.2022.996469
- Schreiner TG, Genes TM. Obesity and Multiple Sclerosis-A Multifaceted Association. Journal of clinical medicine. 2021;10(12):2689. DOI:10.3390/jcm10122689
- Keyhanian K, Saxena S, Gombolay G, Healy BC, Misra M, Chitnis T. Adipokines are associated with pediatric multiple sclerosis risk and course. Multiple sclerosis and related disorders. 2019;36:101384. DOI:10.1016/j.msard.2019.101384
- Chu F, Shi M, Lang Y, et al. Gut Microbiota in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis: Current Applications and Future Perspectives. Mediators of inflammation. 2018;2018:8168717. DOI:10.1155/2018/8168717
- Stoiloudis P, Kesidou E, Bakirtzis C, et al. The Role of Diet and Interventions on Multiple Sclerosis: A Review. Nutrients. 2022;14(6):1150. DOI:10.3390/nu14061150
- Gold R, Montalban X, Haghikia A. Multiple sclerosis and nutrition: back to the future?. Therapeutic advances in neurological disorders. 2020;13:1756286420936165. DOI:10.1177/1756286420936165
- Alkhawajah NM, Aljarallah S, Hussain-Alkhateeb L, Almohaini MO, Muayqil TA. Waterpipe Tobacco Smoking and Other Multiple Sclerosis Environmental Risk Factors. Neuroepidemiology. 2022;56(2):97-103. DOI:10.1159/000521223
- Encinas JM, Manganas L, Enikolopov G. Nitric oxide and multiple sclerosis. Current neurology and neuroscience reports. 2005;5(3):232–238. DOI:10.1007/s11910-005-0051-y
- Iova OM, Marin GE, Lazar I, et al. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders-An Overview. Antioxidants (Basel). 2023;12(3):753.DOI:10.3390/antiox12030753
- Haase S, Linker RA. Inflammation in multiple sclerosis. Therapeutic advances in neurological disorders. 2021;14:17562864211007687. DOI:10.1177/17562864211007687
- Lo J, Chan L, Flynn S. A Systematic Review of the Incidence, Prevalence, Costs, and Activity and Work Limitations of Amputation, Osteoarthritis, Rheumatoid Arthritis, Back Pain, Multiple Sclerosis, Spinal Cord Injury, Stroke, and Traumatic Brain Injury in the United States: A 2019 Update. Archives of physical medicine and rehabilitation. 2021;102(1):115-131. DOI:10.1016/j.apmr.2020.04.001
- Olek MJ. Multiple Sclerosis. Annals of internal medicine. 2021;174(6):ITC81-ITC96. DOI:10.7326/AITC202106150
- Simpson S Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. Journal of neurology, neurosurgery, and psychiatry. 2011;82(10):1132-1141.DOI:10.1136/jnnp.2011.240432
- Ward M, Goldman MD. Epidemiology and Pathophysiology of Multiple Sclerosis. Continuum. 2022;28(4):988-1005. DOI:10.1212/CON.0000000000001136
- Dobson R, Rice DR, D’hooghe M, et al. Social determinants of health in multiple sclerosis. Nature reviews. Neurology. 2022;18(12):723-734. DOI:10.1038/s41582-022-00735-5
- Ontaneda D, Amezcua L. Re-thinking race and geography in multiple sclerosis. Multiple sclerosis (Houndmills, Basingstoke, England). 2024;30(1):16-18. DOI:10.1177/13524585231205969
- Kurtzke JF. Epidemiology in multiple sclerosis: a pilgrim’s progress. Brain. 2013;136(Pt 9):2904-2917. DOI:10.1093/brain/awt220
- Pitt D, Lo CH, Gauthier SA, et al. Toward Precision Phenotyping of Multiple Sclerosis. Neurology(R) neuroimmunology & neuroinflammation. 2022;9(6):e200025. DOI:10.1212/NXI.0000000000200025
- Ontaneda D, Rae-Grant AD. Management of acute exacerbations in multiple sclerosis. Annals of Indian Academy of Neurology. 2009;12(4):264-272.DOI:10.4103/0972-2327.58283
- López-Gómez J, Sacristán Enciso B, Caro Miró MA, Querol Pascual MR. Clinically isolated syndrome: Diagnosis and risk of developing clinically definite multiple sclerosis. Neurologia. 2023;38(9):663-670. DOI:10.1016/j.nrleng.2021.01.010
- Lebrun-Frénay C, Rollot F, Mondot L, et al.Risk Factors and Time to Clinical Symptoms of Multiple Sclerosis Among Patients With Radiologically Isolated Syndrome. JAMA network open. 2021;4(10):e2128271. DOI:10.1001/jamanetworkopen.2021.28271
- Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clinical medicine. 2020;20(4):380-383. DOI:10.7861/clinmed.2020-0292
- Petzold A, Fraser CL, Abegg M, et al.Diagnosis and classification of optic neuritis. The Lancet. Neurology. 2022;21(12):1120-1134. DOI:10.1016/S1474-4422(22)00200-9
- Gelfand JM. Multiple sclerosis: diagnosis, differential diagnosis, and clinical presentation. Handbook of clinical neurology, 2014;122:269–290. DOI:10.1016/B978-0-444-52001-2.00011-X
- Beckmann Y, Özakbaş S, Bülbül NG, et al.Reassessment of Lhermitte’s sign in multiple sclerosis. Acta neurologica Belgica. 2015;115(4):605-608. DOI:10.1007/s13760-015-0466-4
- Rommer PS, Eichstädt K, Ellenberger D, et al. Symptomatology and symptomatic treatment in multiple sclerosis: Results from a nationwide MS registry. Multiple sclerosis. 2019;25(12):1641-1652. DOI:10.1177/1352458518799580
- Spirin NN, Kiselev DV, Karpova MS. Neiropaticheskie bolevye sindromy u patsientov s rasseyannym sklerozom [Neuropathic pain syndromes in patients with multiple sclerosis]. Zh Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova. 2021;121(7. Vyp. 2):22-30. DOI:10.17116/jnevro202112107222
- Broche-Pérez Y, Jiménez-Morales RM, Monasterio-Ramos LO, Vázquez-Gómez LA, Fernández-Fleites Z. Fear of Relapse Scale: Spanish version and psychometric characteristics in a sample of patients with Relapsing-Remitting multiple sclerosis. Neurologia. DOI:10.1016/j.nrleng.2022.06.003
- McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA. 2021;325(8):765-779. DOI:10.1001/jama.2020.26858
- Kantarci OH. Phases and Phenotypes of Multiple Sclerosis. Continuum. 2019;25(3):636-654. DOI:10.1212/CON.0000000000000737
- Kuhlmann T, Moccia M, Coetzee T, et al.Multiple sclerosis progression: time for a new mechanism-driven framework. The Lancet. Neurology. 2023;22(1):78-88. DOI:10.1016/S1474-4422(22)00289-7
- Cree BAC, Arnold DL, Chataway J, et al.Secondary Progressive Multiple Sclerosis: New Insights. Neurology. 2021;97(8):378-388.DOI:10.1212/WNL.0000000000012323
- Amezcua L. Progressive Multiple Sclerosis. Continuum. 2022;28(4):1083-1103. DOI:10.1212/CON.0000000000001157
- Hauser SL, Cree BAC. Treatment of Multiple Sclerosis: A Review. The American journal of medicine. 2020;133(12):1380-1390.e2. DOI:10.1016/j.amjmed.2020.05.0S
- Pérez CA, Cuascut FX, Hutton GJ. Immunopathogenesis, Diagnosis, and Treatment of Multiple Sclerosis: A Clinical Update. Neurologic clinics. 2023;41(1):87-106.DOI:10.1016/j.ncl.2022.05.004
- Polman CH, Reingold SC, Banwell B, et al.Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of neurology. 2011;69(2):292-302. DOI:10.1002/ana.22366
- Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. The Lancet. Neurology. 2018;17(2):162-173. DOI:10.1016/S1474-4422(17)30470-2
- Çinar BP, Yorgun YG. What We Learned from The History of Multiple Sclerosis Measurement: Expanded Disability Status Scale. Noro psikiyatri arsivi. 2018;55(Suppl 1):S69-S75.DOI:10.29399/npa.23343
- Díaz C, Zarco LA, Rivera DM. Highly active multiple sclerosis: An update. Multiple sclerosis and related disorders. 2019;30:215-224. DOI:10.1016/j.msard.2019.01.039
- Cross A, Riley C. Treatment of Multiple Sclerosis. Continuum. 2022;28(4):1025-1051. DOI:10.1212/CON.0000000000001170
- McGinley MP, Goldschmidt CH, Rae-Grant AD. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA. 2021;325(8):765-779. DOI:10.1001/jama.2020.26858
- Talanki Manjunatha R, Habib S, Sangaraju SL, Yepez D, Grandes XA. Multiple Sclerosis: Therapeutic Strategies on the Horizon. Cureus. 2022;14(5):e24895. DOI:10.7759/cureus.24895
- Selmaj K, Cree BAC, Barnett M, Thompson A, Hartung HP. Multiple sclerosis: time for early treatment with high-efficacy drugs. Journal of Neurology. 2024;271(1):105-115. DOI:10.1007/s00415-023-11969-8
Citación recomendada: Guzmán Ríos, E. D., Romero, F., Hernández González, R., Jaramillo, M., Pérez, M., Madera, E., & Olvera Castro, J. F. (2025). Esclerosis múltiple: de la desmielinización a la neurodegeneración. Alerta, Revista científica Del Instituto Nacional De Salud, 8(1), 122–132. https://doi.org/10.5377/alerta.v8i1.19198







{kind=link}
{kind=link}
{kind=link}
{kind=link}