
Resumen
Presentación del caso. Se trata de una paciente femenina de 45 años con antecedentes de hipertensión arterial y múltiples cirugías por cáncer, entre ellas, cáncer de tiroides, carcinoma de parótida, cáncer de mama y cáncer endometrial. De manera incidental se identificó una lesión en el hemisferio cerebeloso derecho en una tomografía de senos paranasales, que fue confirmada a través de una resonancia magnética cerebral. La lesión presentaba una apariencia estriada, característica de gangliocitoma displásico del cerebelo o enfermedad de Lhermitte-Duclos. Considerando los antecedentes de diversos tipos de cáncer y los criterios de diagnóstico propuestos por el Consorcio Internacional Cowden y la Red Nacional Integral del Cáncer, se estableció el diagnóstico de síndrome de Cowden que había pasado desapercibido hasta el momento. Intervención terapéutica. Posteriormente, la paciente fue hospitalizada debido al crecimiento de una masa metastásica en el hemicuello derecho con afectación del plexo braquial, adenopatías cervicales, infraclaviculares y axilares derechas. Evolución clínica. En la actualidad, se encuentra recibiendo tratamiento paliativo con el objetivo de controlar los síntomas y mejorar su calidad de vida, ya que expresó su negativa a someterse a una intervención quirúrgica de resección tumoral.
Lhermitte-Duclos Disease and Cowden Syndrome. A Case Report.
Case presentation. The report is of a 45-year-old female patient with a history of high blood pressure and multiple surgeries for cancer, including thyroid cancer, parotid carcinoma, breast cancer, and endometrial cancer. Incidentally, a lesion in the right cerebellar hemisphere was identified in a tomography of the paranasal sinuses, which was later confirmed in a brain magnetic resonance. The lesion had a striated appearance, characteristic of dysplastic gangliocytoma of the cerebellum or Lhermitte-Duclos disease. Considering the history of various types of cancer and the diagnostic criteria proposed by the International Cowden Consortium and the National Comprehensive Cancer Network, the diagnosis of Cowden syndrome, which had gone unnoticed until now, was established. Treatment. Subsequently, the patient was hospitalized due to the growth of a metastatic mass in the right hemicollar with involvement of the brachial plexus, cervical, infraclavicular, and right axillary lymph nodes. Outcome. She is receiving palliative treatment to control the symptoms and improve her quality of life, since she expressed her refusal to undergo tumor resection surgery.
Introducción
La enfermedad de Lhermitte-Duclos (ELD), también conocida como gangliocitoma displásico del cerebelo, es una rara condición descrita por primera vez por Lhermitte y Duclos en 1920. Desde entonces se han reportado alrededor de 300 casos en la literaturai. Se trata de un tumor benigno de crecimiento lento compuesto por células ganglionares atípicas. Según la clasificación de la Organización Mundial de la Salud en su versión de 2021 para el sistema nervioso central, se clasifica como un tumor mixto glioneuronal grado Iii. Suele presentarse en pacientes de 30 a 50 años, en ambos sexos. Los síntomas clínicos están relacionados con su ubicación en la fosa posterior y pueden incluir dolor de cabeza, náuseas y problemas visualesi-iii.
Es importante destacar que este tipo de gangliocitoma puede surgir de forma aislada o asociada al síndrome de Cowden (SC), hasta en un 35 % de los casosiii. Este síndrome es una enfermedad multisistémica muy rara, que se caracteriza por presentar múltiples hamartomas en diversos tejidos, en particular en la piel y las membranas mucosas, así como en el tracto gastrointestinal, mama, tiroides y cerebro, lo que provoca alto riesgo de neoplasias malignas especialmente en mama, tiroides y endometrioiv. El síndrome de Cowden es causado por mutaciones con pérdida de función en el gen supresor de tumores homólogo de fosfatasa y tensina (PTEN), ubicado en el cromosoma 10q23v. Afecta aproximadamente a uno de cada 200 000 nacimientos, pero se cree que se subestima debido a su penetrancia variablevi. Hasta el 2018, se habían identificado en la literatura únicamente 44 pacientesvii.
El diagnóstico del síndrome de Cowden es eminentemente clínico, basado en los criterios de diagnóstico propuestos por el Consorcio Internacional Cowden y la Red Nacional Integral del Cáncer (NCCN, por sus siglas en inglés: National Comprehensive Cancer Network), que incluyen lesiones mucocutáneas y la presencia de la enfermedad de Lhermitte-Duclos, que se ha establecido como criterio diagnóstico mayorviii,ix.
Presentación del caso
Se trata de una paciente femenina de 45 años de edad, quien había recibido tratamiento, durante los últimos dos años, por endocrinología en un hospital público de tercer nivel de San Salvador, El Salvador. La paciente tenía historia de haber recibido atenciones previas en clínicas privadas y en un hospital público periférico por los diagnósticos de hipertensión arterial y antecedente de varios tipos de neoplasias metacrónicas sin valoración genética, entre ellas, cáncer de tiroides, con tiroidectomía hace 14 años, desarrollando hipotiroidismo e hipoparatiroidismo secundario; carcinoma epidermoide de parótida derecha con resección completa, hace 12 años; carcinoma micropapilar invasivo de mama derecha, con mastectomía derecha hace siete años; y cáncer de endometrio, con una histerectomía hace cinco años. La paciente recibió tratamiento diario con levotiroxina sódica 50 μ, carbonato de calcio 1800 mg, vitamina D3 0,25 μ, irbesartán 150 mg y propranolol 40 mg, todos administrados por vía oral.
Durante un seguimiento rutinario, la paciente expresó que mantenía obstrucción nasal y cefalea ocasional, sin otra sintomatología relevante. En el examen físico se observó a la paciente con buen estado general, los signos vitales se encontraban dentro de los parámetros normales, con presión arterial de 120/70 mmHg, frecuencia cardiaca de 85 latidos por minuto, frecuencia respiratoria de 16 respiraciones por minuto, temperatura corporal de 36,4 °C y saturación de oxígeno en sangre del 98 %. No se encontraron secreciones patológicas en las cavidades nasales, ni dolor a la palpación facial, ni alteraciones neurológicas.
Los resultados de los exámenes de laboratorio (Tabla 1) mostraron niveles bajos de hormona estimulante de la tiroides y niveles elevados de tiroxina libre, por lo que se reajustó la dosis de levotiroxina sódica, disminuyéndola a 25 μ en comparación con la dosis previa de 50 μ. Además, se mantuvieron los medicamentos de uso habitual para controlar las patologías asociadas.
| Exámenes | Resultados en la evaluación rutinaria | Resultados en la evaluación a los seis meses | Valores de referencia |
| Hormona estimulante del tiroides | 0,21 UI/mL | 0,81 UI/mL | 0,34 – 5,60 UI/mL |
| Tiroxina libre | 1,33 ng/dL | 1,59 ng/dL | 0,61 – 1,12 ng/dL |
| Antitiroglobulina | 0,00 UI/mL | – | 0,00 – 115,00 UI/mL |
| Tiroglobulina | 5,11 ng/mL | – | 3 – 42 ng/mL |
| Calcio | 8,44 mg/dL | 5,39 mg/dL | 8,5 – 10,2 mg/dL |
| Hemoglobina | – | 10,00 g/dL | 12 – 16 g/dL |
| Glóbulos blancos | – | 11,20 × 103/μL | 5 – 10 × 103/μL |
| Plaquetas | – | 328 × 103/μL | 150 – 400 × 103/μL |
| * Días de enfermedad. Fuente: Datos obtenidos a partir del expediente clínico. | |||
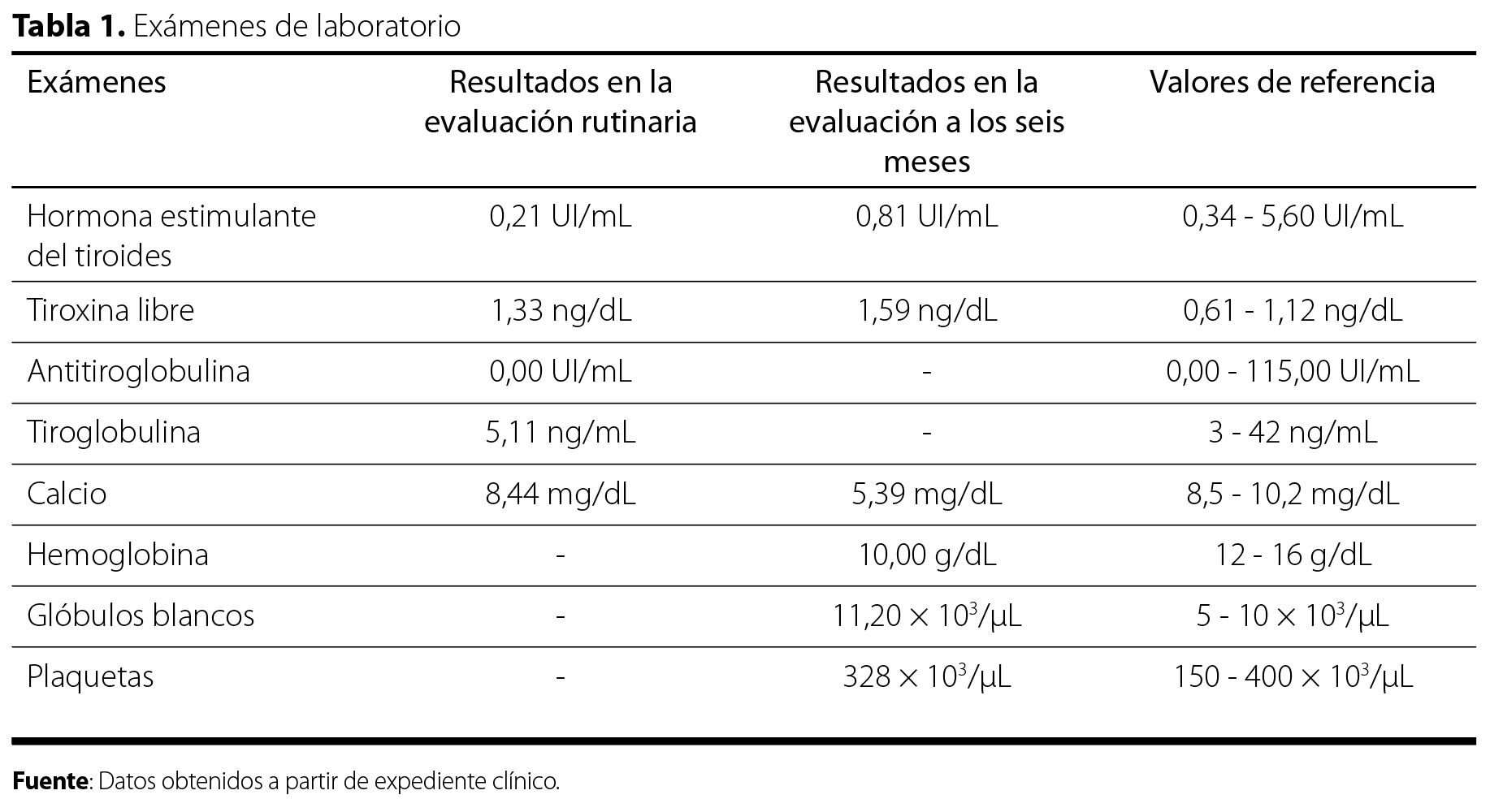
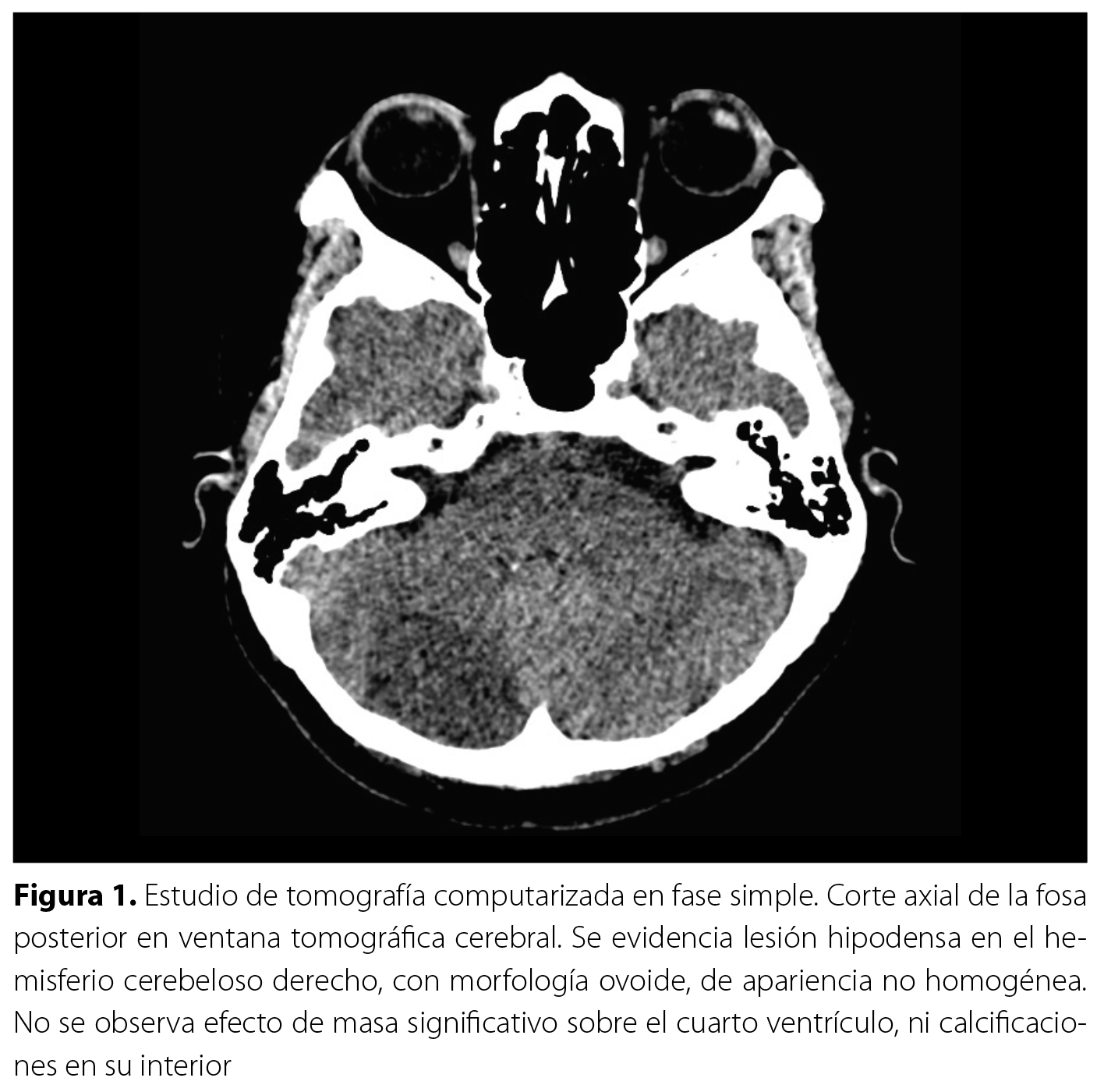
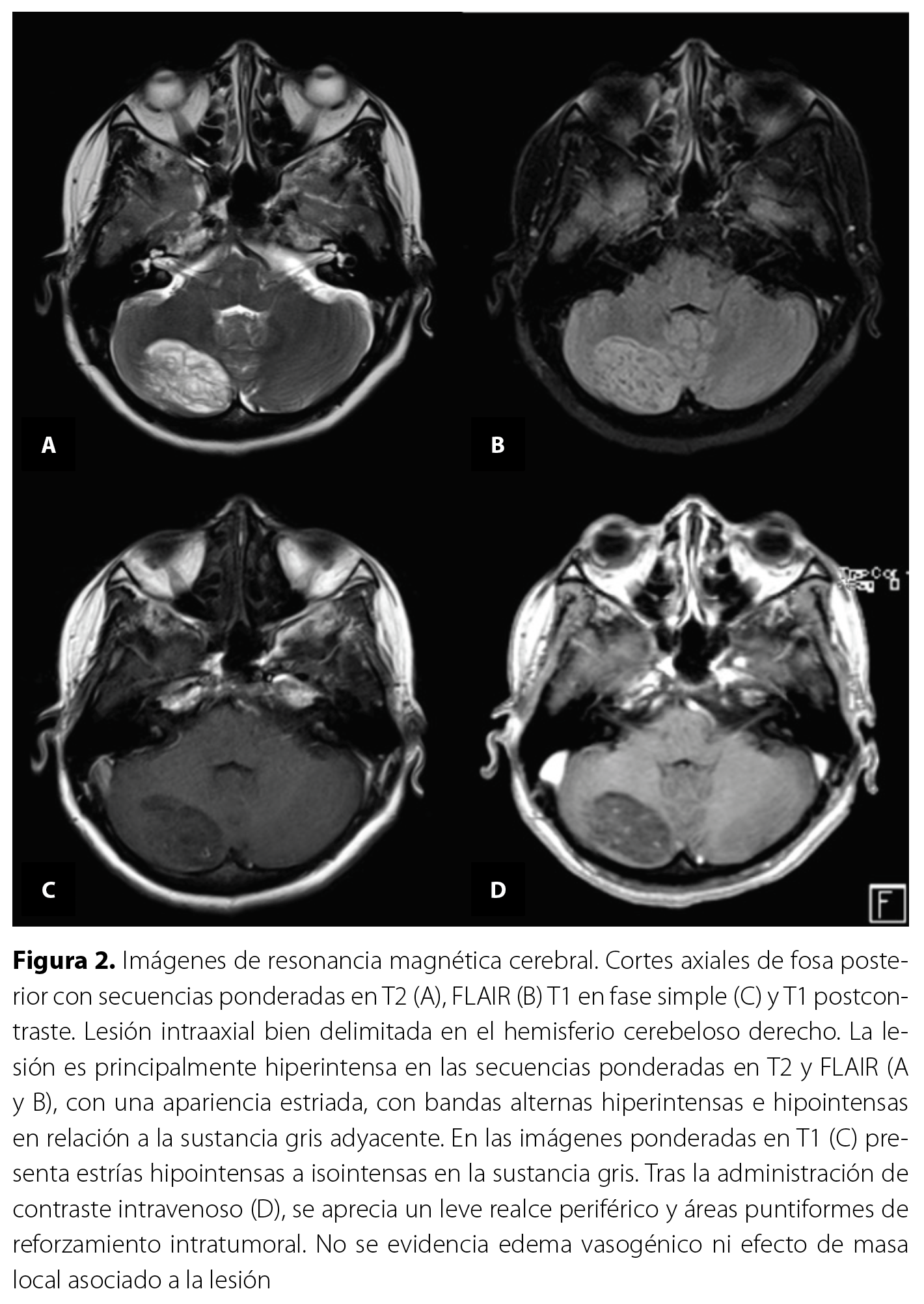
El estudio tomográfico de los senos paranasales describió signos de neumatización adecuada de los senos paranasales y cavidades nasales, sin evidencia de engrosamiento mucoso ni ocupación patológica en los mismos. De manera incidental, se identificó una lesión hipodensa mal definida en el hemisferio cerebeloso derecho, sin características específicas, que no provocaba efecto de masa significativo sobre las estructuras circundantes (Figura 1). Debido a este hallazgo, el Servicio de Radiología recomendó complementar con una resonancia magnética (RM) cerebral. La RM cerebral (Figura 2 y Figura 3) confirmó la presencia de una lesión intraaxial en el hemisferio cerebeloso derecho, con morfología ovoide, contornos regulares y configuración estriada con bandas alternantes hipo e hiperintensas en secuencias ponderadas en T2. No había edema perilesional ni efecto de masa sobre el cuarto ventrículo. Además, mostró un patrón T2 en las secuencias de difusión y posterior a la administración de contraste intravenoso, había un leve realce periférico y áreas puntiformes de reforzamiento intratumoral con dimensiones de 22,3 x 38,8 x 21,8 mm en sus ejes dorsoventral, laterolateral y rostrocaudal. La espectroscopía multivoxel evidenció reducción de N-acetilaspartato y presencia de pico inverso de lactato. Estos hallazgos fueron compatibles con el diagnóstico de gangliocitoma displásico cerebeloso derecho o enfermedad de Lhermitte-Duclos.
Este hallazgo, evaluado en conjunto con los antecedentes de la paciente y basados en las pautas de la NCCN para el diagnóstico de síndrome de Cowden, permitieron identificar el cumplimiento de los criterios mayores, como cáncer de mama, tumor de tiroides, cáncer de útero y enfermedad de Lhermitte-Duclos. Además, de manera intencionada se identificó la presencia de múltiples pápulas en la frente compatibles con triquilemomas, lo que corresponde a otro criterio mayor en la paciente.
Intervención terapéutica
La paciente permaneció asintomática, sin embargo, seis meses posteriores al diagnóstico, consultó por haber notado una masa en el hemicuello derecho, de crecimiento progresivo, que se acompañó de edema, parestesias y parálisis progresiva en el miembro superior derecho. Negó presentar otros síntomas. En el examen físico, se encontró la masa de consistencia sólida, firme al tacto con adherencia a los planos profundos, de aproximadamente 4 × 3 cm. Además, el miembro superior derecho presentaba pérdida de la fuerza muscular. Debido a esto se decidió su ingreso hospitalario.
Los exámenes de laboratorio mostraron valores normales de hormona estimulante de la tiroides y tiroxina libre, niveles bajos de calcio y hemoglobina, así como ligera leucocitosis (Tabla 1).
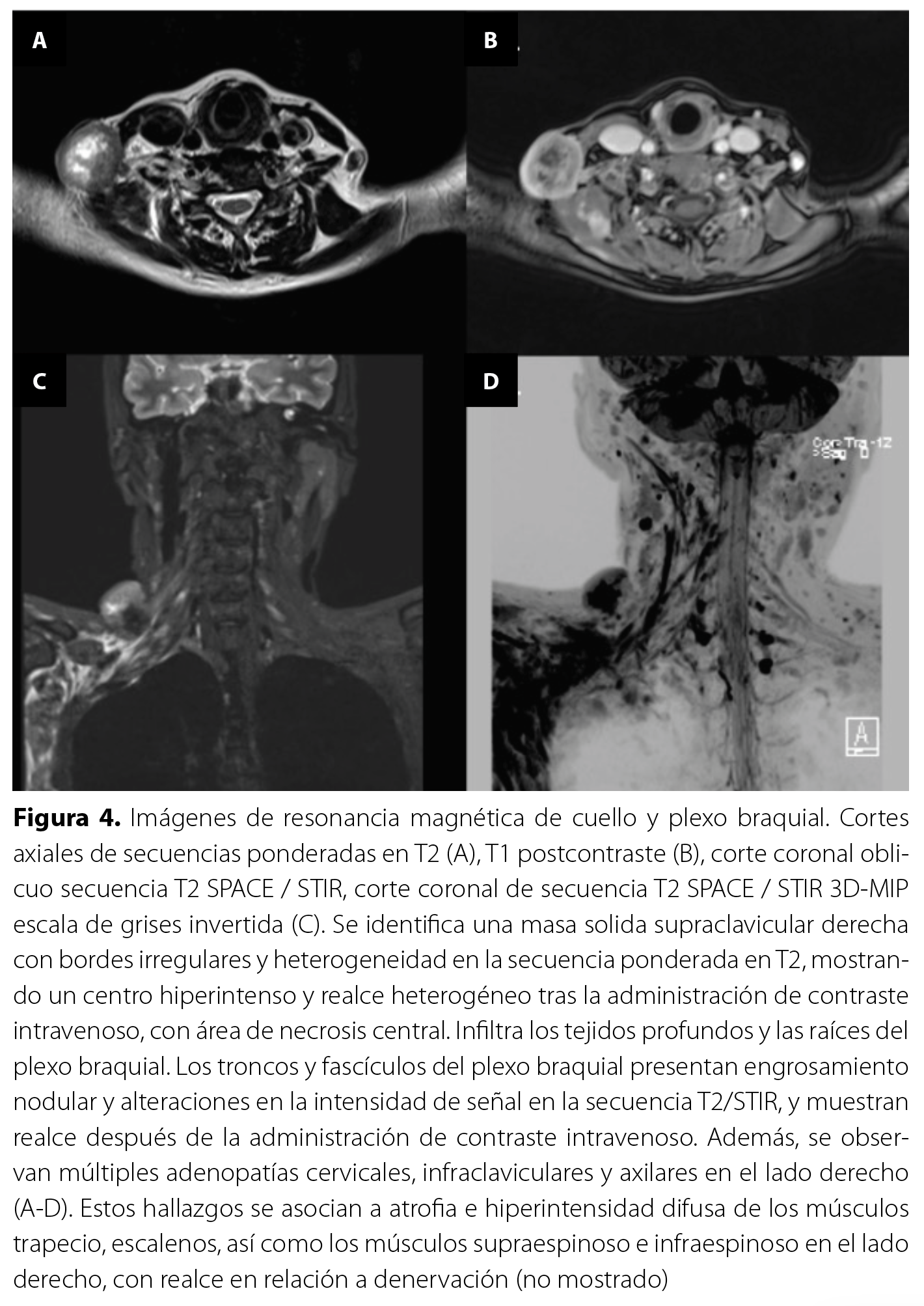
Durante su hospitalización, se realizó una ecografía que reportó una masa heterogénea en la región supraclavicular derecha, así como múltiples adenopatías cervicales, infraclaviculares y axilares derechas. Además, se observó abundante edema de tejido celular subcutáneo en el miembro superior derecho. Debido a ello, se solicitó una RM de cuello y plexo braquial (Figura 4) que confirmó la presencia de la masa en la región supraclavicular derecha, infiltrada en el plexo braquial y los músculos escalenos, trapecio y cintura escapular ipsilaterales, con atrofia muscular y presencia de adenopatías cervicales y axilares derechas. La biopsia de la masa confirmó la presencia de una neoplasia epitelial maligna que se disponía formando nidos y cordones que infiltraban el estroma. El análisis de inmunohistoquímica con panel de mama reportó receptores de estrógenos positivo en el 90 % de células investigadas, receptor de progesterona positivo en el 60 % de células investigadas, HER2: negativo (+/+++), P53: positivo en el 1 % de células investigadas. Ki67: no hay. Los hallazgos podrían corresponder a metástasis por cáncer de mama.
Evolución clínica
La paciente recibió el alta hospitalaria 11 días posteriores a su ingreso, ya que expresó su negativa a someterse a una intervención quirúrgica de resección tumoral. En su última consulta, se brindó tratamiento paliativo para controlar los síntomas y mejorar su calidad de vida. Continuaba con los medicamentos para las enfermedades crónicas de base y se le agregó tramadol 50 mg por vía oral cada ocho horas para el control del dolor.
Debido al patrón de herencia autosómico dominante de esta enfermedad, se brindó consejería genética a los familiares, informándoles sobre el síndrome de Cowden, sus características clínicas y los diferentes tumores asociados. Se destacó la importancia del seguimiento médico regular, que incluye las evaluaciones periódicas y las pruebas de detección específicas de acuerdo con las pautas establecidas.
Diagnóstico clínico
Enfermedad de Lhermitte-Duclos asociado a Síndrome de Cowden (Síndrome de COLD).
Discusión
La enfermedad de Lhermitte-Duclos es una rara lesión hamartomatosa de crecimiento lento en la corteza cerebelosa. Se caracteriza por la presencia de células displásicas en lugar de neoplásicas en el cerebeloi. Se presenta con mayor frecuencia en la tercera y cuarta década de vida. Sin embargo, puede manifestarse en cualquier momento desde el nacimiento hasta la sexta décadai,x.
La naturaleza y patogenia de la ELD aún son motivo de debate. Desde el punto de vista histológico, se caracteriza por engrosamiento y mielinización anormal de la capa molecular en el cerebelo, la atenuación o ausencia de células de Purkinje, la infiltración de la capa de células granulares por células ganglionares displásicas anormales y la variabilidad en la vacuolización de la sustancia blanca. Estos hallazgos histológicos son características distintivas de la enfermedadvii.
Los síntomas característicos incluyen parálisis de nervios craneales, inestabilidad en la marcha, ataxia y deterioro neurológico repentino debido a hidrocefalia aguda o crónicax. La gravedad de los síntomas puede variar según el tamaño de la lesióniii. A medida que el tumor crece, se pueden presentar signos de aumento de la presión intracraneal, como dolores de cabeza, náuseas, vómitos, papiledema, trastornos mentales y pérdida del conocimiento. La duración de los síntomas varía desde unos pocos meses hasta más de diez años. Además, en algunos casos puede presentarse de forma asintomática y ser descubierta de manera incidental durante estudios de imagenxi.
La radiología desempeña un papel importante en el diagnóstico de la ELDiii,x-xiv. La RM se prefiere sobre la tomografía computarizada para evaluar la fosa posterior debido a su capacidad para ofrecer imágenes detalladas de los tejidos blandos y realizar secuencias especializadas que brindan información adicional sobre la estructura y función del cerebelo y el tronco encefálico. En la tomografía computarizada, se observa como una masa cerebelosa hipoatenuada o isoatenuada adyacente al cerebelo normal, sin captación de contraste, y pueden presentarse calcificacionesx,xi.
En las imágenes de RM, se observa que en las secuencias ponderadas en T1 hay una señal hipointensa, mientras que en las secuencias ponderadas en T2 se presenta un patrón alternante de señal alta y baja con una apariencia estriada clásica similar a «rayas de tigre»xi-xiv. Estos hallazgos radiológicos se correlacionan con cambios patológicos en donde el núcleo central de la hipointensidad T1 e hiperintensidad T2 corresponde a sustancia blanca adelgazada, ensanchamiento de la capa de células granulares y las porciones internas de la capa molecular displásica. La capa externa (T1 isointensa, T2 iso a hipointensa) se atribuye a la capa molecular externa y las leptomeninges. En las secuencias de susceptibilidad, pueden observarse vasos anormales y áreas de calcificaciónx.
En las secuencias especiales de RM, específicamente en las secuencias de difusión, se evidencia un efecto T2 en las folias anormalmente engrosadas. En la espectroscopía, se detecta una disminución del N-acetilaspartato y un aumento del lactato, los cuales son rasgos característicosx-xii. Por último, en las imágenes de perfusión, se suele observar una perfusión local elevadaviii.
En este caso, la lesión cerebelosa identificada en la RM mostró la apariencia típica descripta en la literatura médica, que fue relevante para llegar al diagnóstico.
La ELD y el SC están relacionados ya que aproximadamente el 35 % de los pacientes con SC presentan de manera característica el gangliocitoma displásico cerebelosoiii. Esta conexión ha llevado a denominarla síndrome Cowden-Lhermitte-Duclos (síndrome COLD)i,iii. La detección de uno de estos trastornos puede requerir una evaluación y seguimiento más exhaustivos.
El síndrome de Cowden (también conocido como enfermedad de Cowden o síndrome de hamartomas múltiples, OMIM 158350) es un trastorno genético poco común con un patrón de herencia autosómico dominante altamente variable. Descrito por primera vez en 1963 por Lloyd y Dennis, sobre una paciente llamada Rachel Cowdenvi,xv. Se caracteriza por la presencia de múltiples hamartomas de origen ectodérmico, mesodérmico y endodérmicovii. La pérdida de la función del gen PTEN contribuye a un sobrecrecimiento hamartomatoso benigno de los tejidos, especialmente en la piel y las membranas mucosas, así como en el tracto gastrointestinal, mama, tiroides y cerebro, por lo que tienen un mayor riesgo de neoplasias malignas, especialmente cáncer de mama, cáncer de tiroides y cáncer de úteroiv. Es típicamente una enfermedad de adultos jóvenes, que se presenta en la segunda o tercera década de la vida, con una edad promedio de diagnóstico de 39 años (entre 40 a 75 años). Es un poco más común en las mujeresxv.
En 1997 se descubrió que el SC está relacionado con mutaciones en el gen PTEN, presente en el cromosoma 10q23xv. Aproximadamente el 80 % de los pacientes tienen una mutación identificable en este gen. PTEN es un gen supresor de tumores que codifica una fosfatasa y regula negativamente las vías de señalización de PI3K/AKT y mTOR. Controla la proliferación celular, la progresión del ciclo celular y la apoptosis. La pérdida de la función de PTEN contribuye a la transformación celular y aumenta el riesgo de desarrollar cáncer en múltiples órganosiv,xv.
Las manifestaciones benignas incluyen múltiples pólipos gastrointestinales (93 %, con un 44 % como hamartomas), características dermatológicas (98 %), macrocefalia (93 %), lesiones mamarias benignas (74 %), lesiones tiroideas (71 %) y malformaciones vasculares (18 a 35 %)xvi. Los pacientes tienen un riesgo de por vida de cáncer de mama (85 %), tiroides (38 %), endometrio (28 %), colorrectal (9 %) y melanoma (6 %)6,15.
Los criterios de diagnóstico actualizados para el SC fueron desarrollados por Pilarski et al. en 2013viii y reconocidos por la Red Nacional Integral del Cáncer. Según las pautas de la NCCN (Tabla 1)viii,ix, el diagnóstico se basa en la presencia de una variante patógena/probablemente patógena en el gen PTEN y/o criterios clínicos específicos: tres o más criterios mayores (uno de los cuales debe ser macrocefalia, ELD o hamartomas gastrointestinales) o dos criterios mayores y tres menores.
Estas pautas recomiendan realizar una vigilancia por imágenes en pacientes con SC para la detección temprana de posibles cánceres, lo que permite una resección oportuna de las neoplasiasix. Este enfoque de detección intencionada tiene como objetivo identificar y tratar cualquier malignidad de manera temprana, mejorando así el pronóstico y los resultados clínicos en los pacientesxv.
El tratamiento recomendado para la ELD es la observación con control de los síntomas, a menos que los síntomas del efecto de masa sean lo suficientemente problemáticos como para justificar la cirugía. La resección quirúrgica completa se asocia con bajas tasas de recurrenciaiii. Sin embargo, durante la cirugía, el principal desafío técnico es la falta de un margen claro entre el tumor y el tejido cerebral normal. La resección total de la lesión es difícil debido al crecimiento lento del tumor y los límites difusos con el cerebelo adyacentex. Se ha descrito la ecografía intraoperatoria como una herramienta útil para la evaluación en tiempo real durante las operaciones neuroquirúrgicasxiv.
Aspectos éticos
En el caso presentado se ha respetado la confidencialidad de la paciente y se tuvo el consentimiento informado del responsable de la paciente.
Agradecimiento
Al personal del Servicio de Medicina Interna y Radiología e Imágenes del Hospital Nacional Rosales por su contribución en la atención y diagnóstico del caso.
Financiamiento
Los autores declaran no tener fuente de financiamiento.
- Ideta MML, Paschoalino MCO, Oliveira LM, Santos NBD, Sanz MRV, Amorim RLOD. Lhermitte-Duclos Disease and Cowden Syndrome: A Case Report and Literature Review. Arq Bras Neurocir. 2019;38(04):319-323. DOI: 10.1055/s-0039-1693682
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-1251. DOI: 10.1093/neuonc/noab106
- Joo GJ, Doumanian J. Radiographic Findings of Dysplastic Cerebellar Gangliocytoma (Lhermitte-Duclos Disease) in a Woman with Cowden Syndrome: A Case Study and Literature Review. Radiology Case. 2020;14(3):1-6. DOI: 10.3941/jrcr.v14i3.3814
- Morisaki T, Kashiwagi S, Kouhashi R, Yabumoto A, Asano Y, Takashima T, et al. Cowden Syndrome Diagnosed by Bilateral Breast Cancer with Lhermitte-Duclos Disease: A Case Report. Case Rep Oncol. 2020;13(1):419-423. DOI: 10.1159/000506979
- Nelen M. Germline mutations in the PTEN/MMAC1 gene in patients with Cowden disease. Human Molecular Genetics. 1997;6(8):1383-1387. DOI: 10.1093/hmg/6.8.1383
- Montagne W, Wang RC. Cowden Syndrome Case Report: Use of an Ultrasonic Surgical Aspirator for Cosmetic Removal of Lip Hamartomas. Cureus. 2022 Oct 2. DOI: 10.7759/cureus.29839
- Dhamija R, Weindling SM, Porter AB, Hu LS, Wood CP, Hoxworth JM. Neuroimaging abnormalities in patients with Cowden syndrome: Retrospective single-center study. Neur Clin Pract. 2018;8(3):207-213. DOI: 10.1212/CPJ.0000000000000463
- Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden Syndrome and the PTEN Hamartoma Tumor Syndrome: Systematic Review and Revised Diagnostic Criteria. JNCI Journal of the National Cancer Institute. 2013;105(21):1607-1616. DOI: 10.1093/jnci/djt277
- Daly RM, Iuliano S, Fyfe JJ, Scott D, Kirk B, Thompson MQ, et al. Screening, Diagnosis and Management of Sarcopenia and Frailty in Hospitalized Older Adults: Recommendations from the Australian and New Zealand Society for Sarcopenia and Frailty Research (ANZSSFR) Expert Working Group. J Nutr Health Aging. 2022;26(6):637-651. DOI: 10.1007/s12603-022-1801-0
- Cheng CS, Ou CH, Chen JS, Lui CC, Yeh LR. Lhermitte-Duclos disease: A case report with radiologic-pathologic correlation. Radiol Case Rep. 2019;14(6):734-739. DOI: 10.1016/j.radcr.2019.03.020
- Beniwal HK, Satish T, Rao GP, Reddy MS, Manne S. Lhermitte-Duclos Disease: Incidental Finding in Traumatic Cerebral Hemorrhage. Indian Journal of Neurotrauma. 2022;19(01):044-046. DOI: 10.1055/s-0041-1729136
- Zhang H-W, Zhang Y-Q, Liu X-L, Mo Y-Q, Lei Y, Lin F, et al. MR imaging features of Lhermitte-Duclos disease: Case reports and literature review. Medicine (Baltimore). 2022;101(4):e28667. DOI: 10.1097/MD.0000000000028667
- Khandpur U, Huntoon K, Smith-Cohn M, Shaw A, Elder JB. Bilateral Recurrent Dysplastic Cerebellar Gangliocytoma (Lhermitte-Duclos Disease) in Cowden Syndrome: A Case Report and Literature Review. World Neurosurg. 2019;127:319-325. DOI: 10.1016/j.wneu.2019.03.131
- Li Y, Guo J, Wei H, Sun C, Chai Y, Fu X, et al. The surgical resection of dysplastic cerebellar gangliocytoma assisted by intraoperative sonography: illustrative case. Journal of Neurosurgery: Case Lessons. 2021;2(14):CASE21451. DOI: 10.3171/CASE21451
- Dragoo DD, Taher A, Wong VK, Elsaiey A, Consul N, Mahmoud HS, et al. PTEN Hamartoma Tumor Syndrome/Cowden Syndrome: Genomics, Oncogenesis, and Imaging Review for Associated Lesions and Malignancy. Cancers. 2021;13(13):3120. DOI: 10.3390/cancers13133120
- Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50(4):255-263. DOI: 10.1136/jmedgenet-2012-101339
Citación recomendada: Contreras de Montenegro KJ, Fuentes EJ, Sosa ME. Enfermedad de Lhermitte- Duclos y síndrome de Cowden. Reporte de un caso. Alerta. 2024;7(1):29-35. DOI: 10.5377/ alerta.v7i1.16358







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}