
Resumen
La enfermedad de Parkinson y Alzheimer son las enfermedades neurodegenerativas más frecuentes a nivel mundial. Tienen etiología multifactorial, entre ellas, la genética; y son motivo de interés en la investigación científica actual. Se realizó una revisión narrativa con el objetivo de determinar las alteraciones genéticas asociadas a estas patologías, además su influencia en la evolución y respuesta al tratamiento de ellas. Se consultaron artículos originales, revisiones bibliográficas, sistemáticas, metaanálisis en inglés y español, con fecha de publicación entre el 1 enero de 2018 y el 20 de mayo de 2023, en bases como PubMed y Medline. Se utilizaron los términos MeSH «Alzheimer Disease», «Parkinson Disease», «Drug Therapy» y «Mutations». El riesgo hereditario para la enfermedad de Parkinson suele ser poligenético, sin embargo, existen genes relacionados con mutaciones monogénicas. Se identifican alteraciones en genes de α-sinucleína, glucocerebrosidasa y quinasa 2 rica en leucina que se relacionan con mayor riesgo de desarrollar Parkinson, además de variaciones en el cuadro clínico y edad de inicio de síntomas. En cuanto a la enfermedad de Alzheimer, las alteraciones en los genes de la proteína precursora amiloide, presenilina 1 y 2 se relacionan con la forma familiar de la enfermedad; por otra parte, las de apolipoproteína E4 se han identificado en la forma esporádica, por lo que se consideran como el factor de riesgo genético más importante para su desarrollo.
Genetic alterations Associated with Parkinson’s and Alzheimer’s Disease: Evolution and Response to Treatment.
Parkinson's and Alzheimer's are the most frequent neurodegenerative diseases worldwide. They have a multifactorial etiology, including genetics, and are of interest in current scientific research. A narrative review was carried out with the aim of determining the genetic alterations associated with these pathologies, as well as their influence on their evolution and response to treatment. Original articles, literature reviews, systematic reviews, meta-analyses in English and Spanish, with publication date between January 1, 2018 and May 20, 2023, were consulted in databases such as PubMed and Medline. MeSH terms "Alzheimer Disease", "Parkinson Disease", "Drug Therapy" and "Mutation" were used. Hereditary risk for Parkinson's disease is usually polygenetic, however, there are genes related to monogenic mutations. Alterations in α-synuclein, glucocerebrosidase and leucine-rich kinase 2 genes have been identified that are related to an increased risk of developing Parkinson's disease, in addition to variations in the clinical picture and age of symptom onset. As for Alzheimer's disease, alterations in the genes of the amyloid precursor protein, presenilin 1 and 2 are related to the familial form of the disease; on the other hand, those of apolipoprotein E4 have been identified in the sporadic form, and are therefore considered to be the most important genetic risk factor for its development.
Introducción
Entre las enfermedades neurodegenerativas más frecuentes, destacan la enfermedad de Alzheimer (EA) y la enfermedad de Parkinson (EP). La EA es la causa de demencia más frecuente, representando entre el 60 y 80 % de los casos a nivel mundial. La Organización Mundial de la Salud reportó una prevalencia de 8,5 millones de casos de EP para el año 2019, duplicando su prevalencia en los últimos 25 añosi,ii.
La neurogenética es el campo de la ciencia que estudia la función de los genes en el desarrollo y función del sistema nerviosoiii. Las intervenciones en neurogenética están dirigidas a la identificación de los procesos fisiopatológicos primarios que comienzan años antes de que aparezcan los síntomasiv.
Se han descrito múltiples variantes en la presentación de enfermedades neurodegenerativas, dependiendo de la edad en que se presentan, clasificándose de inicio temprano o tardío. La importancia de caracterizar estas variantes genotípicas radica en la posibilidad de identificar la predisposición genética de algunos pacientes, que pueden debutar con síntomas atípicos y agresivos, con un peor pronóstico, de forma más oportuna. Esto brinda la oportunidad de identificar nuevos biomarcadores, dianas terapéuticas, y la elaboración de escalas de riesgo para estas patologíasv.
En esta revisión narrativa se realizó una búsqueda de información de artículos originales y revisiones bibliográficas, sistemáticas, metaanálisis en inglés y español, en las bases de datos PubMed, Medline y SciELO, usando los términos MeSH «Alzheimer Disease», «Parkinson Disease», «Drug Therapy» y «Mutations», publicados principalmente entre el 1 de enero de 2018 y 20 de mayo de 2023, incluyendo además algunos artículos de años anteriores que se consideraron relevantes para la base de esta revisión. Por lo tanto, esta revisión pretende determinar las alteraciones genéticas asociadas a las enfermedades de Parkinson y Alzheimer; y su influencia en la evolución y respuesta al tratamiento de estas patologías.
Discusión
Alteraciones genéticas relacionadas a la enfermedad de Parkinson
Se estima que entre 3 y 5 % de casos de EP están vinculados a alteraciones en genes conocidos con riesgo hereditario monogénico, y entre 16 y 36 % a riesgo hereditario no monogénico, describiéndose más de 90 variantes involucradasvi; Nalls et al. realizaron un estudio de asociación del genoma completo, en el que compararon 37 688 casos de EP con 1,4 millones de controles, identificando 78 loci que afectan el riesgo de EP. Sus hallazgos sugieren que las variantes identificadas con mayor frecuencia presentan menor riesgo patogénico, pero al interactuar con factores genéticos y ambientales contribuyen al grado de riesgo de EP. Sin embargo, mutaciones monogénicas en los genes de la α-sinucleína (SNCA), quinasa 2 repetida rica en leucina (LRRK2), parkin (PRKN), quinasa 1 inducida por PTEN (PINK1), y glucocerebrosidasa (GBA), generan riesgo elevadovii.
Para el gen SNCA, en el cromosoma 4q, que codifica la proteína α-sinucleína, se han identificado mediante estudios de secuenciación variantes con significado indeterminado en la EP autosómica dominante; Day et al., sugieren que dichas mutaciones producen mal plegamiento de la proteína, posterior agregación acelerada y acumulación intracelular, incrementando el estrés oxidativo mediante interrupción de la función del proteosomaviii.
El gen LRRK2, situado en el cromosoma 12q, codifica la enzima quinasa, donde sus mutaciones se asocian con EP autosómica dominante, identificándose en aproximadamente 1 a 2 % de todos los pacientes con EP y en el 5 % de la forma familiarix. Jankovic et al. describen que los puntos críticos de mutación están ubicados principalmente en los dominios funcionales de la enzima, generando desregulación de las actividades de la quinasa y la GTPasa, con una ganancia tóxica de función que podría ser el mecanismo subyacentex.
Las mutaciones en los genes PRKN y PINK1, ubicados en los cromosomas 6q y 1p respectivamente, se han identificado hasta en 77 % de los casos de EP juvenil y entre 10 a 20 % de los casos de inicio temprano. Poseen herencia autosómica recesiva; generando lisosomas de mitocondrias disfuncionales por macroautofagia, lo que produce mitofagia alterada. Simon et al., describen que PARKIN regula indirectamente niveles del PGC-1α-, importante regulador transcripcional, involucrado en expresión de genes necesarios para biogénesis mitocondrial y múltiples defensas antioxidantesvi,ix.
El gen GBA, ubicado en el cromosoma 1q, codifica la enzima lisosomal glucocerebrosidasa, esta descompone el glucocerebrósido en glucosa y ceramida, importante en la degradación de esfingolípidos. Las variantes de este ocurren aproximadamente en el 8,5 % de los pacientes con EP y patrón hereditario autosómico dominante; los portadores tienen un riesgo aproximadamente cuatro veces mayor de EP que la población general, asociado a acumulación y agregación de α-sinucleínax.
Enfermedad de Alzheimer
Para la EA de inicio temprano (cuando los síntomas se desarrollan previo a los 65 años), se han identificado más de 400 mutaciones en tres genes: proteína precursora de amiloide (APP), presenilina-1 (PSEN1) y presenilina-2 (PSEN2); que representan 5 % de casos de EA con patrón hereditario autosómico dominante. La EA de inicio tardío (cuando los síntomas se desarrollan posterior a los 65 años), se asocia a polimorfismos del gen de apolipoproteína E (Apo E), presentes hasta en 65 % de los casos, siendo el principal gen de susceptibilidadii.
El gen de la APP, ubicado en el cromosoma 21q, codifica una proteína transmembrana de la cual derivan los β-amiloide (Aβ) por acción de gamma-secretasas; Kamboh et al., describen que su mutación representa 10 % al 15 % de casos de EA familiar autosómica dominante; afectando la función de la gamma secretasa y aumentando la producción de Aβxi.
Los genes PSEN1 y PSEN2, ubicados en cromosomas 14q y 1q, respectivamente, codifican las proteínas PSEN1 y PSEN2, las cuales son parte del complejo gamma-secretasa que regulan la actividad proteolítica de gamma-secretasa sobre APP. Breijyeh et al., describen que sus mutaciones en su mayoría tienen significado indeterminado con patrón de herencia autosómico dominantexii.
Qin et al. postulan que, la mutación de PSEN1 es frecuente hasta en 75 % de casos de EA familiar, con más de 200 variaciones en el mismo; en cambio en PSEN2, se presentan en cerca del 12 % con más de 40 variaciones; con alteración de la función de gamma-secretasa, produciendo Aβ; las mutaciones de PSEN1 alteran la función neuronal afectando la actividad de GSK-3β y la motilidad basada en kinesina-I, lo que lleva a neurodegeneraciónxiii.
En el cromosoma 19 se ubica el gen de Apo E, glicoproteína que se expresa en astrocitos y microglias, en tres isoformas: Apo E2, Apo E3 y Apo E4; esta proteína actúa como ligando de endocitosis mediada por receptores para lipoproteínas, entre ellos el colesterol, importante para la producción de mielina y función cerebral normal; por otra parte, los portadores del alelo Apo E4 tienen mayor riesgo de desarrollar EA, aumentando tres veces para portadores heterocigotos y 15 veces en homocigotos; mientras que la homocigosidad del alelo Apo E2, se ha identificado como factor protector para EAxiv.
Apo E4 se une competitivamente a los receptores Aβ en la superficie de los astrocitos, impidiendo la captación de Aβ, promueve la siembra y agregación de Aβ en oligómeros y fibrillas, reduciendo su eliminación del líquido intersticial. Estas deposiciones de Aβ en forma de placas amiloides, causan angiopatía amiloide cerebral y daño vascular cerebral, importante en la patogénesis de EAxv.
Relación de las alteraciones genéticas con el curso clínico y pronóstico de pacientes
Enfermedad de Parkinson
El cuadro clínico de la EP se caracteriza por síntomas motores y no motores, siendo los síntomas clásicos temblor en reposo, rigidez, bradicinesia y alteraciones posturales. Los síntomas no motores descritos son deterioro cognitivo, disfunción autonómica de tipo ortostatismo, hiperhidrosis, depresión, ansiedad, demencia, trastornos del sueño y anomalías sensoriales como anosmia, parestesias y dolorxvi.
Post et al. definen la EP de inicio temprano al aparecimiento de los síntomas entre los 21 y 40 años de edad, describen que suele haber alteraciones genéticas asociadas con diferencias al curso clínico de EP clásica, con mayor frecuencia de distonías y disquinesias asociadas al uso de levodopa. El subtipo clásico de EP de inicio tardío se caracteriza por aparecimiento de los síntomas después de los 60 añosxvii.
En el «UK Tracking Parkinson Study» Malek eet al., estudiaron a 1893 pacientes con EP y encontraron que la mutación L444P fue la mutación patogénica más frecuente del gen de la GBA. Los pacientes con esta mutación eran en promedio cinco años más jóvenes al inicio de la EP que los no portadores, con más probabilidad de presentar dificultad en la marcha, inestabilidad postural sin diferencias significativas en la función cognitiva en etapas tempranas de la enfermedad comparado con los no portadoresxviii.
Un metanálisis realizado por Creese et al. encontró que los pacientes con EP portadores de mutaciones en GBA tienen 2,4 veces mayor riesgo de desarrollar deterioro cognitivo, además de 1,8 y 2,2 veces mayor riesgo de presentar psicosis y depresión respectivamente, comparado con pacientes con EP esporádico (no portadores de mutaciones)xix.
Las variantes en LRRK2 también se han asociado a cambios en el fenotipo clínico de los pacientes con EP. La presencia de la mutación G2019S se asocia con deterioro motor más lento comparado con pacientes con EP no portadores de la mutación. Yahalom et al. evaluaron 225 pacientes judíos asquenazíes con EP y encontraron que la mutación G2019S se asoció a menor edad de aparecimiento de síntomas comparado con pacientes con mutación N370S o pacientes sin mutacionesxx.
Omer et al. estudiaron 10 090 pacientes con EP y compararon pacientes con mutaciones en LRRK2, GBA y LRRK2+GBA, identificando que portadores de LRRK2 sola o LRRK2+GBA tienen un fenotipo clínico más leve con mejores puntajes en la escala de evaluación unificada de la EP, comparados con quienes tienen mutaciones solo en GBAxxi.
Pang et al. describen que las mutaciones en LRRK2 modifican el efecto de las mutaciones de GBA, lo que resulta en EP con síntomas más leves, comparado con pacientes con mutaciones solamente en GBA. Sin embargo, los portadores de ambas mutaciones tienen mucho más riesgo para desarrollar EP y menor edad de aparecimiento de los síntomas que los pacientes que solo portan una mutaciónxxii.
Las alteraciones en el gen de la SNCA también confieren cambios en el fenotipo clínico de los pacientes con EP. Magistrelli et al. determinaron que mutaciones en SNCA presentan menor edad de aparecimiento de síntomas, síntomas no motores más severos y más tempranoxxiii. Un metaanálisis conducido por Shu et al., encontró que algunas variantes en SNCA del alelo 271-bp confieren mayor riesgo presentar síntomas a menor edad, mientras que las variantes del alelo 267-bp tienen el efecto opuestoxxiv.
La EP con mutaciones en PRKN se caracteriza por inicio de la enfermedad a edades más tempranas, distonía de miembros inferiores al momento de la presentación, ausencia de deterioro cognitivo, frecuentes fluctuaciones motoras y discinesiasxxv. Por otro lado, el fenotipo de EP con mutaciones en PINK1 se caracteriza por progresión más lenta con síntomas típicos de temblor, bradicinesia, rigidez, menor edad de aparecimiento de síntomas. El deterioro cognitivo y alteraciones psiquiátricas como psicosis son raras en estos pacientesxxvi.
Enfermedad de Alzheimer
La EA se caracteriza clínicamente por un deterioro cognitivo progresivo con alteraciones en memoria episódica de inicio insidioso y de manera progresiva, funciones visuales/espaciales y ejecutivas; la presentación más frecuente es a edad avanzada (mayor de 60 años). Posteriormente surgen dificultades topográficas y dificultades con multitareas, además de cambios de comportamiento, problemas de movilidad, alucinaciones y convulsionesxxvii.
El gen Apo E4 es considerado el factor de riesgo genético más importante para EA esporádica. Tellechea et al. encontraron que la presencia de Apo E4 se asocia a menor edad de inicio de síntomas de EA, más frecuencia de tipo amnésica en comparación con el patrón de preservación en el hipocampoxxviii.
Baril et al. encontraron asociación entre Apo E4 y mayor severidad de insomnio, que empeora la función cognitiva y memoria en EAxxix. Frey et al. estudiaron 144 pacientes con EA y encontraron que quienes no portan el alelo E4 del Gen Apo E presentan fenotipo no amnésico de EA, muestran deterioro cognitivo en dominios no relacionados con la memoria (lenguaje, comportamiento, atención, funciones ejecutivas y visuoespaciales)xxx.
Las alteraciones en el PSEN1 se asocian a un promedio de edad menor en el aparecimiento de los síntomas comparado con las alteraciones en los genes de APP y PSEN2xxxi. Huang eet al. encontraron que la mutación Asp678His en el gen de la APP se asoció a una progresión más rápida a demencia severa en 5 a 10 años de la edad de inicio de los síntomasxxxii. Wang et al. encontraron que los pacientes con la mutación Ile716Thr en la APP presentaron un marcado deterioro en la memoria situacional, con una edad de inicio de los síntomas entre los 35 y 40 añosxiii.
Al estudiar la mutación V717I de la APP en cinco familias chinas, Zhang et al. encontraron que la edad media de aparecimiento de los síntomas fue 54,7 años, los síntomas iniciales fueron disfunción ejecutiva, desorientación y pérdida sutil de la memoria; los síntomas neurológicos fueron de aparición tardía y se caracterizaron por marcada paraparesia espástica y ataxia cerebelosaxxxiii.
Qiu et al. encontraron una nueva alteración genética (Gly111Val) en el gen de la PSEN1; no encontraron diferencias en el fenotipo clínico de los portadores, siendo la pérdida de memoria a corto plazo el síntoma más frecuentexxxiv. En cambio, Li et al. estudiaron la variante Glu116Lys, y encontraron que los portadores presentaron desorientación y deterioro en la memoria desde los 35 años y progresaron rápidamente con síntomas psiquiátricos; algunos pacientes murieron a los 40 añosxxxv.
Otro estudio conducido por Qiu et al. encontró la alteración M139L en el PSEN1 y reporta que la edad media de inicio de los síntomas fue de 45 años, los principales síntomas que experimentaron los portadores fueron deterioro progresivo de la memoria, alteraciones visuoespaciales e irritabilidadxxxvi. Li et al., también encontraron la variante Ile202Phe, cuyos portadores mostraron deterioro en la memoria desde los 36 años y posteriormente desarrollaron dificultades con el lenguaje y cambios en la personalidadxxxvii.
Las mutaciones en PSEN2 suelen ser más raras. Qin et al. encontraron que los pacientes con estas mutaciones tienen una edad de aparecimiento de los síntomas mayor que los pacientes con mutaciones en APP o PSEN1, tienen una progresión más lenta y síntomas similares a la EA esporádica o idiopáticaxiii.
Relación de las alteraciones genéticas con la respuesta al tratamiento
Enfermedad de Parkinson
En la EP, la levodopa sigue siendo el estándar de oro en el manejo inicial, con mayor beneficio en control de las manifestaciones motoras en comparación con agonistas dopaminérgicos; dos vías importantes se ven implicadas en la síntesis de dopamina, la vía del catecol o-metiltransferasa y la amino oxidasa Bxxxviii.
Las descripciones iniciales de LRRK2 enfatizaron las similitudes clínicas entre esta condición y la EP idiopática, específicamente que ambas son formas progresivas de parkinsonismo que responden bien a la terapia con levodopa (L-DOPA)xxxix.
Lantin et al. llevaron a cabo un estudio para explorar la asociación entre los polimorfismos en los genes de las vías dopaminérgicas, encontrando que el polimorfismo rs921451 en el gen dopa-decarboxilasa tuvo efecto sobre la respuesta al tratamiento con L-DOPA en pacientes chinos con EP. Sin embargo, esto podría variar debido a las diferencias étnicasxl.
Así mismo las mutaciones asociadas a las vías dopaminérgicas tienen efecto importante en los efectos adversos de los fármacos. Yin et al. evidenciaron en su metaanálisis que el genotipo AA del polimorfismo rs4680 de la catecol-O-metiltransferasa aumenta potencialmente el riesgo de disquinesia inducida por levodopa en un modelo genético recesivo para pacientes con EPxli.
Enfermedad de Alzheimer
La Administración de Alimentos y Medicamentos ha aprobado dos grupos farmacológicos para el tratamiento de EA, los inhibidores de la acetilcolinesterasa (CEI), entre ellos el donepezilo, rivastigmina y galantamina. Otro grupo son los moduladores de los receptores N-metil-D-aspartato, el único aprobado es la memantinaxlii. Estos no actúan a nivel de los procesos patológicos involucrados en el desarrollo de la enfermedad, además las diferentes variantes genéticas son responsables aproximadamente del 75 a 85 % de la variabilidad en la respuesta al tratamientoxliii.
Cheng et al., en un metaanálisis que incluyó 30 estudios, no identificaron influencia significativa en la respuesta al tratamiento con CEI en pacientes portadores del alelo Apo E4 en comparación con los no portadoresxliv. Jia et al. llevaron a cabo un estudio multicéntrico prospectivo con 241 pacientes en el que identificaron mejor respuesta al tratamiento por parte de pacientes no portadores, con incremento en la puntuación del Mini-Mental State Examinationxlv.
De igual forma se ha identificado mejor respuesta al tratamiento con donepezilo o rivastigmina en pacientes portadores de la variante Apo E3 en comparación a Apo E4xlvi. En pacientes que además presentan el genotipo BCH K, se evidencia menor respuesta a rivastigmina y a memantina, debido a la sinergia existente entre el alelo Apo E4 con dicha variantexlvii.
Wallin et al., en un estudio multicéntrico prospectivo, identificaron mejor respuesta al tratamiento con galantamina en pacientes mayores con capacidades cognitivas y funcionales bajas al inicio del estudio, tasa de progresión previa al tratamiento más rápida y menor incidencia del alelo Apo E4xlviii.
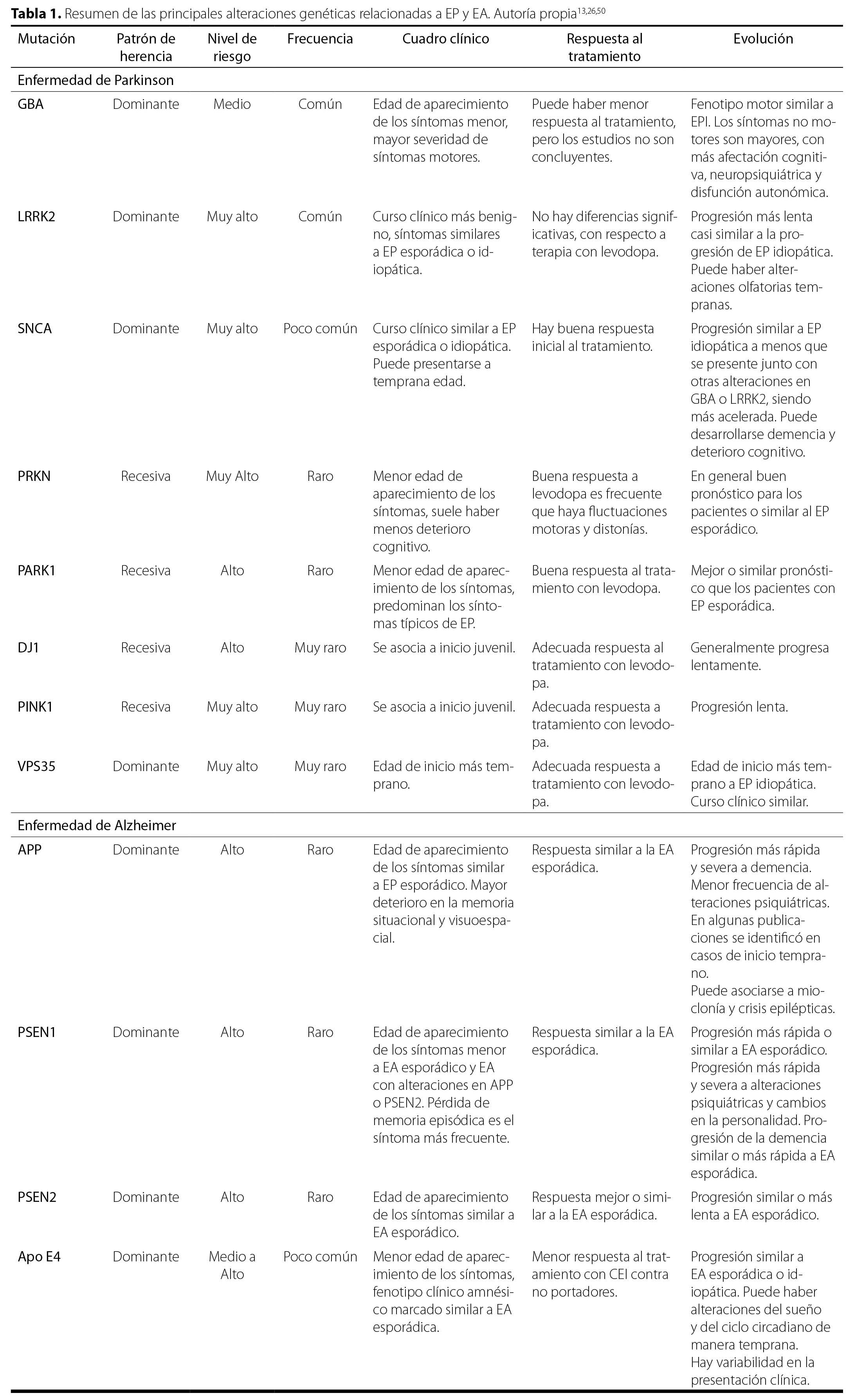
Los fármacos para pacientes con EA que tienen mutación de PSEN1 se limitan a terapias sintomáticas y no hay tratamientos específicos disponibles. La reutilización de fármacos basada en células madre pluripotentes inducidas identificó a la bromocriptina como candidato terapéutico para EA con mutación de PSEN1xlix. En la Tabla 1 se resumen las principales alteraciones genéticas estudiadas en esta revisión y algunas menos frecuentes, pero también de interés en la comunidad científica.
Actualmente, la evidencia sigue en construcción. Se están realizando estudios para evaluar la influencia de la genética en la clínica y respuesta a tratamientos específicos de personas con enfermedades neurodegenerativas como EP y EA.
| Mutación | Nivel de riesgo | Frecuencia | Cuadro clínico | Respuesta al tratamiento | Evolución | |
| Enfermedad de Parkinson | ||||||
| GBA | Dominante | Medio | Común | Edad de aparecimiento de los síntomas menor, mayor severidad de síntomas motores. | Puede haber menor respuesta al tratamiento, pero los estudios no son concluyentes. | Fenotipo motor similar a EPI. Los síntomas no motores son mayores, con más afectación cognitiva, neuropsiquiátrica y disfunción autonómica. |
| LRRK2 | Dominante | Muy alto | Común | Curso clínico más benigno, síntomas similares a EP esporádica o idiopática. | No hay diferencias significativas, con respecto a terapia con levodopa. | Progresión más lenta casi similar a la progresión de EP idiopática. Puede haber alteraciones olfatorias tempranas. |
| SNCA | Dominante | Muy alto | Poco común | Curso clínico similar a EP esporádica o idiopática. Puede presentarse a temprana edad. | Hay buena respuesta inicial al tratamiento. | Progresión similar a EP idiopática a menos que se presente junto con otras alteraciones en GBA o LRRK2, siendo más acelerada. Puede desarrollarse demencia y deterioro cognitivo. |
| PRKN | Recesiva | Muy Alto | Raro | Menor edad de aparecimiento de los síntomas, suele haber menos deterioro cognitivo. | Buena respuesta a levodopa es frecuente que haya fluctuaciones motoras y distonías. | En general buen pronóstico para los pacientes o similar al EP esporádico. |
| PARK1 | Recesiva | Alto | Raro | Menor edad de aparecimiento de los síntomas, predominan los síntomas típicos de EP. | Buena respuesta al tratamiento con levodopa. | Mejor o similar pronóstico que los pacientes con EP esporádica. |
| DJ1 | Recesiva | Alto | Muy raro | Se asocia a inicio juvenil. | Adecuada respuesta al tratamiento con levodopa. | Generalmente progresa lentamente. |
| PINK1 | Recesiva | Muy alto | Muy raro | Se asocia a inicio juvenil. | Adecuada respuesta a tratamiento con levodopa. | Progresión lenta. |
| VPS35 | Dominante | Muy alto | Muy raro | Edad de inicio más temprano. | Adecuada respuesta a tratamiento con levodopa. | Edad de inicio más temprano a EP idiopática. Curso clínico similar. |
| Enfermedad de Alzheimer | ||||||
| APP | Dominante | Alto | Raro | Edad de aparecimiento de los síntomas similar a EP esporádico. Mayor deterioro en la memoria situacional y visuoespacial. | Respuesta similar a la EA esporádica. | Progresión más rápida y severa a demencia. Menor frecuencia de alteraciones psiquiátricas. En algunas publicaciones se identificó en casos de inicio temprano. Puede asociarse a mioclonía y crisis epilépticas. |
| PSEN1 | Dominante | Alto | Raro | Edad de aparecimiento de los síntomas menor a EA esporádico y EA con alteraciones en APP o PSEN2. Pérdida de memoria episódica es el síntoma más frecuente. | Respuesta similar a la EA esporádica. | Progresión más rápida o similar a EA esporádico. Progresión más rápida y severa a alteraciones psiquiátricas y cambios en la personalidad. Progresión de la demencia similar o más rápida a EA esporádica. |
| PSEN2 | Dominante | Alto | Raro | Edad de aparecimiento de los síntomas similar a EA esporádico. | Respuesta mejor o similar a la EA esporádica. | Progresión similar o más lenta a EA esporádico. |
| Apo E4 | Dominante | Medio a Alto | Poco común | Menor edad de aparecimiento de los síntomas, fenotipo clínico amnésico marcado similar a EA esporádica. | Menor respuesta al tratamiento con CEI contra no portadores. | Progresión similar a EA esporádica o idiopática. Puede haber alteraciones del sueño y del ciclo circadiano de manera temprana. Hay variabilidad en la presentación clínica. |
Conclusión
Se han descrito variaciones genéticas que influyen en la edad de presentación de síntomas en enfermedades neurodegenerativas, con edades de presentación menor a la evolución usual, además de síntomas neurocognitivos y motores más discapacitantes. En EP se observan edades más tempranas de aparición de síntomas en pacientes con mutaciones de los genes GBA, SNCA, PRKN y PARK1.
En cuanto a respuesta a tratamientos, en pacientes con mutaciones de GBA se ha descrito menor respuesta con progresión más rápida a déficit cognitivo. En EA se desarrolla demencia a menor edad en personas con mutaciones del gen PSEN1. Se ha observado menor respuesta a tratamientos convencionales con mutaciones de APO E4. La progresión es más rápida y severa en mutaciones del gen APP y PSEN1.
- Organización Mundial de la Salud. Enfermedad de Parkinson. 2023. Fecha de consulta: 9 de agosto de 2023. Disponible en: https://www.who.int/es/news-room/fact-sheets/detail/parkinson-disease
- Andrade J, Santiago A, Aguilar J, Vargas I, Cadena A, Sánchez C, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. IJMS. 2023;24(4):3754. DOI: 10.3390/ijms24043754
- Izquierdo A, Palomo M, Celi J. Neurogenética. Medicine – Programa de Formación Médica Continuada Acreditado. 2019;12(77):4559-4566. DOI: 10.1016/j.med.2019.05.004
- Smedinga M, Darweesh S, Bloem B, Post B, Richard E. Towards early disease modification of Parkinson’s disease: a review of lessons learned in the Alzheimer field. J Neurol. 2021;268(2):724-733. DOI: 10.1007/s00415-020-10162-5
- Mendez M. Early-onset Alzheimer Disease and Its Variants. CONTINUUM: Lifelong Learning in Neurology. 2019;25(1):34-51. DOI: 10.1212/CON.0000000000000687
- Bloem B, Okun M, Klein C. Parkinson’s disease. The Lancet. 2021;397(10291):2284-2303. DOI: 10.1016/S0140-6736(21)00218-X
- Nalls M, Blauwendraat C, Vallerga CL, Heilbron K, Bandres S, Chang D, et al. Expanding Parkinson’s disease genetics: novel risk loci, genomic context, causal insights and heritable risk. Genetics; 2018. DOI: 10.1101/388165
- Day J, Mullin S. The Genetics of Parkinson’s Disease and Implications for Clinical Practice. Genes. 2021;12(7):1006. DOI: 10.3390/genes12071006
- Simon D, Tanner C, Brundin P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clinics in Geriatric Medicine. 2020;36(1):1-12. DOI: 10.1016/j.cger.2019.08.002
- Jankovic J, Tan E. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795-808. DOI: 10.1136/jnnp-2019-322338
- Kamboh M. Genomics and Functional Genomics of Alzheimer’s Disease. Neurotherapeutics. 2022;19(1):152-172. DOI: 10.1007/s13311-021-01152-0
- Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. 2020;25(24):5789. DOI: 10.3390/molecules25245789
- Qin Q, Yin Y, Wang Y, Lu Y, Tang Y, Jia J. Gene mutations associated with early onset familial Alzheimer’s disease in China: An overview and current status. Molec Gen & Gen Med. 2020;8(10):e1443. DOI: 10.1002/mgg3.1443
- Hoogmartens J, Cacace R, Van Broeckhoven C. Insight into the genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rare variants. Alz & Dem Diag Ass & Dis Mo. 2021;13(1):e12155. DOI: 10.1002/dad2.12155
- Serrano A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. The Lancet Neurology. 2021;20(1):68-80. DOI: 10.1016/S1474-4422(20)30412-9
- Beitz J. Parkinson s disease a review. Front Biosci. 2014;S6(1):65-74. DOI: 10.2741/S415
- Post B, Van Den Heuvel L, Van Prooije T, Van Ruissen X, Van De Warrenburg B, Nonnekes J. Young Onset Parkinson’s Disease: A Modern and Tailored Approach Bloem BR, Brundin P, editors. JPD. 2020;10(s1):S29-S36. DOI: 10.3233/JPD-202135
- Malek N, Weil R, Bresner C, Lawton M, Grosset K, Tan M, et al. Features of GBA -associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J Neurol Neurosurg Psychiatry. 2018;89(7):702-709. DOI: 10.1136/jnnp-2017-317348
- Creese B, Bell E, Johar I, Francis P, Ballard C, Aarsland D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: Review and meta-analyses. American J of Med Genetics Pt B. 2018;177(2):232-241. DOI: 10.1002/ajmg.b.32549
- Yahalom G, Rigbi A, Israeli S, Krohn L, Rudakou U, Ruskey J, et al. Age at Onset of Parkinson’s Disease Among Ashkenazi Jewish Patients: Contribution of Environmental Factors, LRRK2 p.G2019S and GBA p.N370S Mutations. JPD. 2020;10(3):1123-1132. DOI: 10.3233/JPD-191829
- Omer N, Giladi N, Gurevich T, Bar A, Gana M, Goldstein O, et al. A Possible Modifying Effect of the G2019S Mutation in the LRRK2 Gene on GBA Parkinson’s Disease. Movement Disorders. 2020;35(7):1249-1253. DOI: 10.1002/mds.28066
- Pang S, Lo RCN, Ho PW-L, Liu H-F, Chang EES, Leung C-T, et al. LRRK2, GBA and their interaction in the regulation of autophagy: implications on therapeutics in Parkinson’s disease. Transl Neurodegener. 2022;11(1):5. DOI: 10.1186/s40035-022-00281-6
- Magistrelli L, Contaldi E, Comi C. The Impact of SNCA Variations and Its Product Alpha-Synuclein on Non-Motor Features of Parkinson’s Disease. Life. 2021;11(8):804. DOI: 10.3390/life11080804
- Shu L, Zhang Y, Sun Q, Pan H, Guo J, Tang B. SNCA REP1 and Parkinson’s disease. Neuroscience Letters. 2018;682:79-84. DOI: 10.1016/j.neulet.2018.05.043
- Lesage S, Lunati A, Houot M, Romdhan S, Clot F, Tesson C, et al. Characterization of Recessive Parkinson Disease in a Large Multicenter Study. Annals of Neurology. 2020;88(4):843-850. DOI: 10.1002/ana.25787
- Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt E, et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin , PINK1, DJ1: MDSGene Systematic Review. Movement Disorders. 2018;33(5):730-741. DOI: 10.1002/mds.27352
- Aoki Y, Takahashi R, Suzuki Y, Pascual R, Kito Y, Hikida S, et al. EEG resting-state networks in Alzheimer’s disease associated with clinical symptoms. Sci Rep. 2023;13(1):3964. DOI: 10.1038/s41598-023-30075-3
- Tellechea P, Pujol N, Esteve P, Echeveste B, García M, Arbizu J, et al. Enfermedad de Alzheimer de inicio precoz y de inicio tardío: ¿son la misma entidad? Neurología. 2018;33(4):244-253. DOI: 10.1016/j.nrl.2015.08.002
- Baril A, Beiser A, Sanchez E, Mysliwiec V, Redline S, Gottlieb DJ, et al. Insomnia symptom severity and cognitive performance: Moderating role of APOE genotype. Alzheimer’s & Dementia. 2022;18(3):408-421. DOI: 10.1002/alz.12405
- Fray S, Achouri A, Belal S, Messaoud T. Missing apolipoprotein E ɛ4 allele associated with nonamnestic Alzheimer’s disease in a Tunisian population. J Genet. 2022;101(2):41. DOI: 10.1007/s12041-022-01384-9
- Wu L, Rosa P, Hsiung G, Sadovnick A, Masellis M, Black S, et al. Early-Onset Familial Alzheimer’s Disease (EOFAD). Can. J. Neurol. Sci. 2012;39(4):436-445. DOI: 10.1017/S0317167100013949
- Huang C, Hsiao I, Lin K, Huang K, Fung H, Liu C, et al. Amyloid PET pattern with dementia and amyloid angiopathy in Taiwan familial AD with D678H APP mutation. Journal of the Neurological Sciences. 2019;398:107-116. DOI: 10.1016/j.jns.2018.12.039
- Zhang G, Xie Y, Wang W, Feng X, Jia J. Clinical characterization of an APP mutation (V717I) in five Han Chinese families with early-onset Alzheimer’s disease. Journal of the Neurological Sciences. 2017;372:379-386. DOI: 10.1016/j.jns.2016.10.039
- Qiu Q, Jia L, Wang Q, Zhao L, Jin H, Li T, et al. Identification of a novel PSEN1 Gly111Val missense mutation in a Chinese pedigree with early-onset Alzheimer’s disease. Neurobiology of Aging. 2020;85:155.e1-155.e4. DOI: 10.1016/j.neurobiolaging.2019.05.018
- Dongxiao Li, Yupeng Liu, Yuan Ding, Xiyuan Li, Xun Wu, Jinqing Song, et al. A patient with initial symptom of epilepsy from age of 12 years old due to early-onset Alzheimer disease caused by presenilin 1 mutation. Chinese Journal of Obstetrics and Gynecology, 2016;12(6):621-626. DOI: 10.3877/CMA.J.ISSN.1673-5250.2016.06.001
- Qiu Q, Shen L, Jia L, Wang Q, Li F, Li Y, et al. A Novel PSEN1 M139L Mutation Found in a Chinese Pedigree with Early-Onset Alzheimer’s Disease Increases Aβ42/Aβ40 ratio. JAD. 2019;69(1):199-212. DOI: 10.3233/JAD-181291
- Li Y, Yang Z, Zhang Y, Yang J, Shang D, Zhang S, et al. Two Novel Mutations and a de novo Mutation in PSEN1 in Early-onset Alzheimer’s Disease. Aging and disease. 2019;10(4):908. DOI: 10.14336/AD.2018.1109
- Pringsheim T, Day G, Smith D, Rae A, Licking N, Armstrong M, et al. Dopaminergic Therapy for Motor Symptoms in Early Parkinson Disease Practice Guideline Summary: A Report of the AAN Guideline Subcommittee. Neurology. 2021;97(20):942-957. DOI: 10.1212/WNL.0000000000012868
- Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16(2):97-107. DOI: 10.1038/s41582-019-0301-2
- Li L, Lin H, Hua P, Yan L, Dong H, Li T, et al. Polymorphism of the Dopa-Decarboxylase Gene Modifies the Motor Response to Levodopa in Chinese Patients With Parkinson’s Disease. Front. Neurol. 2020;11:520934. DOI: 10.3389/fneur.2020.520934
- Yin Y, Liu Y, Xu M, Zhang X, Li C. Association of COMT rs4680 and MAO-B rs1799836 polymorphisms with levodopa-induced dyskinesia in Parkinson’s disease—a meta-analysis. Neurol Sci. 2021;42(10):4085-4094. DOI: 10.1007/s10072-021-05509-3
- Argueta N, Notari E, Szigeti K. Role of Pharmacogenomics in Individualizing Treatment for Alzheimer’s Disease. CNS Drugs. 2022;36(4):365-376. DOI: 10.1007/s40263-022-00915-3
- Zúñiga T, Yescas P, Fricke I, González M, Ortega A, López M. Estudios farmacogenéticos en la enfermedad de Alzheimer. Neurología. 2022;37(4):287-303. DOI: 10.1016/j.nrl.2018.03.025
- Cheng Y, Huang Y, Liu H. Effect of Apolipoprotein E ɛ4 Carrier Status on Cognitive Response to Acetylcholinesterase Inhibitors in Patients with Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Dement Geriatr Cogn Disord. 2018;45(5-6):335-352. DOI: 10.1159/000490175
- Jia J, Wei C, Chen W, Jia L, Zhou A, Wang F, et al. Safety and Efficacy of Donepezil 10 mg/day in Patients with Mild to Moderate Alzheimer’s Disease. Journal of Alzheimer's Disease,. 2020;74(1):199-211. DOI: 10.3233/JAD-190940
- Cacabelos R. Pharmacogenetic considerations when prescribing cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opinion on Drug Metabolism & Toxicology. 2020;16(8):673-701. DOI: 10.1080/17425255.2020.1779700
- Sonali N, Tripathi M, Sagar R, Velpandian T, Subbiah V. Clinical Effectiveness of Rivastigmine Monotherapy and Combination Therapy in Alzheimer’s Patients. CNS Neurosci Ther. 2013;19(2):91-97. DOI: 10.1111/cns.12036
- Wallin Å, Minthon, Wattmo C. Galantamine treatment in Alzheimer’s disease: response and long-term outcome in a routine clinical setting. NDT. 2011;7(1):565-576. DOI: 10.2147/NDT.S24196
- Kondo T, Banno H, Okunomiya T, Amino Y, Endo K, Nakakura A, et al. Repurposing bromocriptine for Aβ metabolism in Alzheimer’s disease (REBRAnD) study: randomised placebo-controlled double-blind comparative trial and open-label extension trial to investigate the safety and efficacy of bromocriptine in Alzheimer’s disease with presenilin 1 (PSEN1) mutations. BMJ Open. 2021;11(6):e051343. DOI: 10.1136/bmjopen-2021-051343
- Yat-Fung Shea, Leung-Wing Chu, Angel On-Kei Chan, Joyce Ha, Yan Li, You-Qiang Song. A systematic review of familial Alzheimer’s disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. Journal of the Formosan Medical Association. 2016;115(2):67-75. DOI: 10.1016/j.jfma.2015.08.004
Citación recomendada: Quezada Rivera RA, Bonilla Rodríguez FE, Benavides Romero MA, Peña Martínez SL. Alteraciones genéticas asociadas a la enfermedad de Parkinson y Alzheimer: evolución y respuesta al tratamiento. Alerta. 2024;7(1):79-87. DOI: 10.5377/ alerta.v7i1.16684







{kind=link}