
Resumen
Presentación del caso. Paciente fenotípicamente femenina de 18 años de edad, cariotipo 46 XY, que fue evaluada en centro de atención de tercer nivel de medicina pediátrica, por un cuadro de amenorrea primaria. En la evaluación física se encontró un adecuado desarrollo mamario, genitales externos femeninos con leve hipotrofia de labios mayores y menores, canal vaginal de 11 cm; ultrasonido reporta útero rudimentario lineal e imágenes sugestivas a ovarios. La resonancia magnética describe presencia de estructura de aspecto tubular bilobulada compuesta por dos imágenes nodulares, las cuales se ubican adyacentes a la pared vesical en su aspecto posterior, lateral y superior derecho de 27,4 × 15,4 × 11,0 mm. El conducto inguinal izquierdo presenta en dos imágenes: una nodular sólida, ovoide, isointensa de 37 × 21 × 13,8 mm (nodular), quística 22,7 × 14,0 mm, ausencia congénita de útero. Intervención terapéutica. En la exploración laparoscópica se detectó presencia de gónadas en canal inguinal y paravesical. Se procedió a la resección de ambas gónadas. Evolución clínica. Se manejó con analgesia, antibiótico y fue dada de alta a los tres días posteriores a la cirugía; la biopsia reportó criptorquidia de testículo derecho e izquierdo.
Presentation of the case. Phenotypically female patient, 18 years old, 46 XY karyotype, who was evaluated in a tertiary care center for pediatric medicine due to primary amenorrhea. Physical evaluation found adequate breast development, female external genitalia with slight hypoatrophy of the labia majora and minora, vaginal canal of 11 cm, ultrasound reports a linear rudimentary uterus and images suggestive of ovaries, magnetic resonance describes the presence of a bilobed tubular structure composed of two nodular images, which are located adjacent to the bladder wall in their aspect posterior, lateral and superior right 27,4 × 15,4× 11,0 mm, left inguinal canal two images, one solid nodular, ovoid, isointense 37 × 21 × 13,8 mm (nodular), cystic 22,7 × 14,0 mm, congenital absence of uterus. Treatment. In the laparoscopic examination, the presence of gonads in the inguinal and para-bladder canals was detected, and both gonads were resected. Outcome. She was managed with analgesia, antibiotics and was discharged three days after surgery. The biopsy reported cryptorchidism of the right and left testis.
Introducción
El síndrome de insensibilidad a los andrógenos (SIA), conocido también como un síndrome de feminización testicular1, incluye un grupo variado de mutaciones que se relacionan con la disfunción de los receptores de andrógenos2 y la resistencia de los tejidos diana a la acción de las hormonas masculinas3. Es causado por alteraciones genéticas localizadas en la secuencia de codificación de los receptores de andrógenos ligada al cromosoma Xq11 - 123, el gen que codifica al receptor de los andrógenos, de un individuo genéticamente masculino (46 XY)3,4. Las formas clinicas moderada, parcial o completa, dependen del grado de insensibilidad androgénica3.
Los avances en la causas genéticas han permitido que estas condiciones congénitas de desarrollo del sexo cromosómico, gonadal o anatómico atípico sean denominados trastornos de diferenciación sexual5.
La característica principal es la resistencia de los tejidos diana a la acción de las hormonas masculinas5,6. La presencia de la proteína de diferenciación sexual Y promueve la formación de testículos primordiales en el abdomen fetal. A partir de la séptima semana después de la concepción, los testículos fetales comienzan a producir testosterona, cuya actividad es bloqueada por los procesos patológicos que afectan a los receptores de andrógenos7, lo que impide el desarrollo masculino normal de los genitales internos y externos de los individuos genéticamente varones6,8 y genera la diferenciación de los genitales externos como femeninos; no se desarrollan los derivados del conducto de Wolff, que dependen de la acción androgénica. El epidídimo, los conductos deferentes y las vesículas seminales9, la presencia de la hormona antimülleriana producida por los testículos primordiales, suprime la formación de los órganos genitales femeninos. La parte inferior de la vagina, en cambio, está completamente desarrollada porque no es un derivado del conducto de Müller. Es más corta de lo normal y presenta un final ciego, carecen de útero y trompas de Falopio7, estructuras müllerianas ausentes o vestigiales y testículos localizados en labios, canal inguinal o abdomen7.
El SIA es una de las causas comunes de los trastornos del desarrollo sexual que dan lugar a fenotipos variados4. Se ha calculado una prevalencia de dos a cinco casos por cada 100 000 varones nacidos genéticos, con una incidencia estimada entre un caso por cada 20 000 a 90 000 varones nacidos genéticos5,10,11.
El espectro fenotípico de los individuos depende de la actividad residual del receptor de andrógenos y abarca desde individuos con un fenotipo completamente femenino, con testículos en ausencia de derivados de los conductos de Wolff y de Müller y ausencia de vello sexual hasta un fenotipo masculino con infertilidad y desvirilización2,3,12. Estas variaciones definen la clasificación en SIA completo y parcial2.
El SIA completo fue descrito por Morris en 1953. Este se caracteriza por presentarse en una persona de apariencia femenina, con desarrollo mamario normal, los genitales externos femeninos de aspecto normal, escaso vello corporal, amenorrea primaria y testículos no descendidos ubicados en lugar de los ovarios1; forma parte de trastornos de diferenciación sexual más frecuentes.
Presentación del caso
Se trata de una paciente fenotípicamente femenina, quien consultó en un hospital de atención infantil a los 16 años de edad por un cuadro de amenorrea, con desarrollo mamario y de genitales externos normales. Con el antecedente quirúrgico de hernioplastia inguinal bilateral a los tres años de edad, en el que reportaron como hallazgo incidental la presencia de ovarios en sacos herniarios, por lo que introdujeron nuevamente a cavidad abdominal. No había iniciado la actividad sexual; telarquia a los 12 años, aparecimiento de vello púbico a los 13 años, no aparecimiento de vello axilar y con preferencia sexual heterosexual. No presentaba otros antecedentes médicos patológicos.
Se indicaron estudios por la sospecha de una amenorrea primaria. Los exámenes de laboratorio reportaron alteraciones hormonales relacionadas con la anovulación, con una elevación leve de las hormonas folículo estimulante y de la luteinizante con niveles bajos de testosterona (Tabla 1). La ultrasonografía pélvica reportó que el útero era escasamente visible, de tamaño infantil, sin evidencia de endometrio; el ovario derecho de 5 cm3 y el izquierdo de 3,9 cm3; no evidenciaron folículos. Se realizó el cariotipo que reportó 46 XY. Fue evaluada por urología pediátrica, donde se indicó una video laparoscopía diagnóstica. En este procedimiento se tomó una muestra de la aparente gónada derecha para una biopsia. El estudio histopatológico reportó un tejido constituido por múltiples focos de túbulos seminíferos hipoplásicos dispersos en estroma ovárico con células de la granulosa, diagnóstico histopatológico ovotestis.
Por llegar a la mayoría de edad fue referida a la unidad de endocrinología de otro hospital, con diagnóstico de trastorno del desarrollo sexual XY, probable síndrome de insensibilidad completa a andrógenos.
En el hospital de referencia fue evaluada clínicamente y se indicaron nuevamente los estudios de laboratorio, los cuales reportaron mantener la elevación leve de las hormonas folículo estimulante, luteinizante y niveles bajos de testosterona (Tabla 1). Además, la ultrasonografía pélvica describió el útero rudimentario lineal 3,7 cm, con imágenes sugestivas de ovarios. Se solicitó repetir la revisión de las láminas de la biopsia anterior y se reportó un tejido testicular constituido por túbulos seminíferos poco desarrollados, algunos con esclerosis y fibrosis de la membrana basal tubular dispuestos en su estroma fibroso, no espermatogénesis, no presencia de tejido ovárico, diagnóstico hipoplasia testicular.
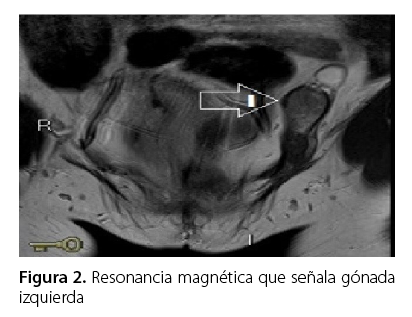
La resonancia magnética abdomino-pélvica describió la presencia de una estructura de aspecto tubular bilobulada, compuesta por dos imágenes nodulares, las cuales se ubicaban adyacentes a la pared vesical en su aspecto posterior, lateral y superior derecho de 27,4 × 15,4 × 11,0 mm (Figura 1). El conducto inguinal izquierdo reflejó dos imágenes nodulares como una sólida, ovoide, isointensa de 37 × 21 × 13,8 mm (nodular) y otra de características quística de 22,7 × 14,0 mm (Figura 2) y ausencia congénita de útero.
Fue evaluada por la unidad de ginecología, donde se le indicó el inicio de la terapia de sustitución hormonal, luego del tratamiento quirúrgico. Además, se refirió al área de atención psicológica donde se desarrolló una sesión junto a la madre de la paciente para evaluar su respuesta al diagnóstico y tratamiento. En esta se concluyó que no era necesario el reforzamiento de género.
La evaluación clínica de la especialidad de urología reportó que el desarrollo mamario era adecuado, con estadio de Tanner V. Se evidenciaron las cicatrices umbilical y parainguinal bilateral, sin otras anormalidades abdominales ni del canal inguinal. También describió los genitales externos femeninos con presencia de vello púbico fino escaso, distribución de vello púbico genital en estadio Tanner V, leve hipotrofia de labios mayores y menores, el himen de forma anular, al realizar el tacto vaginal se identificó el canal vaginal amplio y funcional que solamente permitió la introducción de un dedo, la medición del canal vaginal resultó en 11 cm de longitud. La visualizacióin del meato uretral fue dificil.
Intervención terapéutica
Se programó intervención quirúrgica siete días posterior a evaluación. Se ingresó a la paciente al servicio de endocrinología tres días antes de su intervención quirúrgica para completar las evaluaciones prequirúrgicas. Se indicó la profilaxis con cefazolina de un gramo por vía intravenosa, 30 minutos antes del inicio de la cirugía.
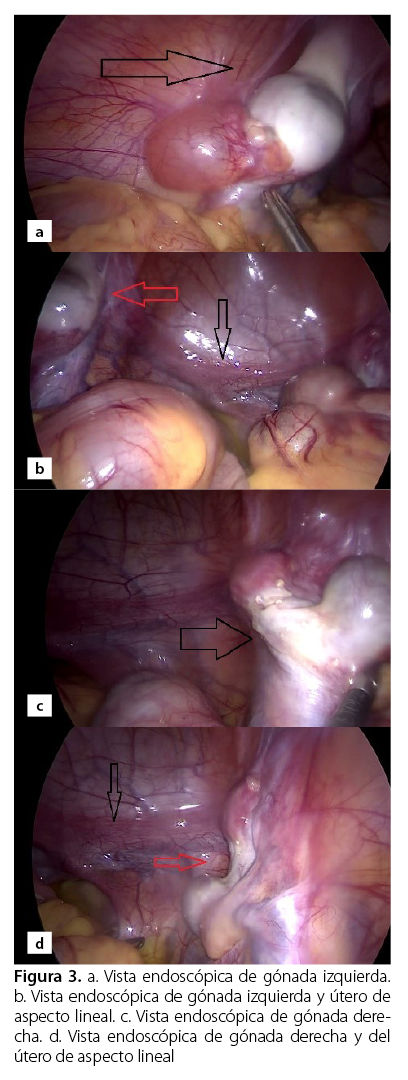
El procedimiento quirúrgico consistió en la extirpación laparoscópica de las gónadas mediante una técnica cerrada. A través de una incisión periumbilical se colocó un trocar de 10 mm; se formó el neumoperitoneo de 12 mmHg y un flujo inicial de 5 L/min. Se colocó un trocar 5 mm en flancos inferiores izquierdo y derecho. Se evidenció la presencia de gónadas a nivel de canal inguinal izquierdo bilobulada (Figura 3), gónada paravesical derecha bilobulada (Figura 3) y el útero de aspecto lineal. Finalmente, se realizó la gonadectomía bilateral y se envió para estudio histológico.
Evolución clínica
En el periodo posquirúrgico inmediato, la paciente recibió tratamiento con ketorolaco y ceftriaxona por vía intravenosa e inició la alimentación seis horas posteriores a cirugía. Se indicó el egreso hospitalario en el tercer día posquirúrgico. Luego de cuatro días, se dio seguimiento para evaluación posquirúrgica y para el reporte de la biopsia que mostró estroma fibromuscular vascularizado con tejido testicular. Presentó túbulos seminíferos sin lumen compuestos únicamente por células de Sertoli; el intersticio presentaba hiperplasia de células de Leydig (Figura 4), sin presencia de epidídimo, de conducto deferente ni de malignidad. Además, se encuentran túbulos pequeños revestidos por epitelio columnar que probablemente correspondía a remanentes paramesonéfricos.
Diagnóstico clínico
Las evaluaciones clínicas de imágenes e histológicas permitieron definir el diagnóstico de síndrome de insensibilidad completa a andrógenos o síndrome de Morris.
Discusión
En el SIA completo no se tiene respuesta a los andrógenos, lo que impide el desarrollo de cualquier característica masculina y se produce un desarrollo genital externo femenino normal12. Es por ello que se educan como mujeres y que su identidad e inclinación sexual no se ven afectadas. Además, sus gónadas son testículos azoospérmicos de localización variable, se encuentran con mayor frecuencia en los conductos inguinales, con presencia o no de epidídimo6.
Es poco probable que se diagnostique durante la infancia. La detección de una hernia inguinal o de signos de edema en los labios mayores en edad preescolar, debe hacer sospechar este síndrome13. La asociación entre la hernia inguinal en niñas prepuberales se ha conocido desde hace más de 60 años. Médicos que tratan las hernias inguinales en la infancia pueden tener la primera oportunidad de diagnosticarlo6. En estos casos, el diagnóstico lo realiza el patólogo tras analizar la pieza quirúrgica13. Lo habitual es diagnosticarlo durante la pubertad, por amenorrea primaria, e incluso más tarde, cuando consulta por esterilidad a un endocrinólogo o ginecólogo.
La pubertad acontece sin virilización y el riesgo de desarrollar un tumor testicular de células germinales13,14 es del 0,02 % en menores de 30 años; por arriba de esa edad, el riesgo aumenta a un 22 %5,7. Por lo que se ha recomendado gonadectomía profiláctica posterior a la pubertad, cuando la feminización de la persona afectada haya completado, ya que se produce en parte por la producción estrogénica testicular y por la conversión periférica de andrógenos a estrógenos. Solo en casos en que los testículos se palpen en la zona inguinal y ocasionen incomodidad o por razones estéticas, será necesaria su extirpación antes de la pubertad, con terapia de remplazo hormonal para iniciar la pubertad hasta alcanzar el desarrollo mamario y genital deseado10.
Posteriormente, se deben iniciar estrógenos a dosis suficientes para permitir la lubricación vaginal y la ganancia ósea5,10. Para las vaginas cortas pueden utilizarse técnicas de dilatación o cirugía que permitan las relaciones sexuales y eviten la dispareunia. Deben mantenerse controles periódicos en las áreas de endocrinología, ginecología, psicología y urología9,10,15. Morris describe que posterior a la castración se presentan sofocos, resequedad vaginal y atrofia de mamas, por lo que no era recomendable realizar este procedimiento1.
Las mujeres que no aceptan la gonadectomía deben mantener un seguimiento anual, debido al riesgo de degeneración maligna, con ultrasonido pélvico y resonancia magnética abdomino-pélvico para evaluar el tamaño y localización de las gónadas, además de la verificación de los marcadores tumorales como alfa feto proteína, gonadotropina coriónica humana fracción beta y lactato deshidrogena16.
Se ha descrito en mujeres con desarrollo mamario normal y de altura elevada, de pelo femenino y sin calvicie, que con menos frecuencia presentan cierta masculinización de genitales externos, como clitoromegalia o fusión de los labios. En algunas de las revisiones, incluso, se ha llegado a encontrar restos de conductos de Müller (trompas de Falopio) hasta en un tercio de las afectadas10.
Los niveles de testosterona se elevan en el periodo de la pubertad, al mismo tiempo que aumentan los niveles de la hormona folículo estimulante con disminución de los niveles de hormona luteinizante lo que sugiere que existe una resistencia androgénica a nivel hipotalámico hipofisiario17. El aumento de las gonadotrofinas, como consecuencia de la insensibilidad, produce un incremento en la producción de testosterona y estradiol por las células de Leydig5,17. De manera análoga, debido a que está conservada la actividad aromatasa (enzima que transforma la testosterona en estradiol), aumenta la síntesis de estrógenos, responsables del desarrollo mamario durante la pubertad de estos individuos5.
Los estudios de imágenes recomendados para confirmar el diagnóstico de ausencia de estructuras müllerianas y localización de las gónadas son la ultrasonografía pélvica y la resonancia magnética13,18. Los testículos se retienen bilateralmente en abdomen entre el 50 y el 70 % de los casos; en la región inguinal, en el 20 %; ubicados uno en región inguinal y otro en abdomen, entre el 10 y el 30 % de los casos. Otras localizaciones poco frecuentes ocurren en la región labioescrotal8.
Los diagnósticos diferenciales a tomar en cuenta corresponden al déficit de la enzima 5α-reductasa, el síndrome de Mayer-Rokintansky-Küster-Hauser, el síndrome de Kallmann, la disgenesia gonadal pura 46 XX, 46 XY, la agenesia de células de Leydig por anomalía en el receptor para la hormona luteinizante y el déficit enzimático que se expresan como ambigüedad sexual al nacimiento18.
En el SIA completo, las características personales y los genitales externos corresponden a los de una mujer. Por lo general, estas mujeres no difieren de otras con respecto a patrones de matrimonio o de otro tipo de relaciones, por lo que no es frecuente que se presenten dilemas de asignación de sexo, aunque algunas de ellas pueden presentar conflictos de identidad sexual19,20. De ahí la importancia de la atención multidiciplinaria, e incluir la colaboración de un psicólogo o psiquiatra en las consideraciones de tratamiento, para evaluar la necesidad de la paciente en el reforzamiento de la identidad sexual; a la familia, a prepararlos para las secuencias de diagnósticos y tratamientos que les esperan durante la vida9.
Aspectos éticos
Para la publicación de este caso se obtuvo el consentimiento informado de la paciente y de la responsable, tanto para la atención hospitalaria en la que se realizó el examen físico como de la publicación de este artículo, con el compromiso de mantener la privacidad de la paciente, según lo establecido en la declaración de Helsinki.
Financiamiento
Autores declaran no tener fuente de financiamiento.
Agradecimiento
Al personal de los servicios de endocrinología y de patología del Hospital Nacional Rosales por su contribución en la atención y diagnóstico del caso.
- Morris JM. The syndrome of testicular feminization in male pseudohermaphrodites. American Journal of Obstetrics and Gynecology. 1953;65(6):1192-1211. DOI: 10.1016/0002-9378(53)90359-7
- Boehmer ALM, Brüggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MCT, Niermeijer MF, et al. Genotype Versus Phenotype in Families with Androgen Insensitivity Syndrome. The Journal of Clinical Endocrinology & Metabolism. 2001;86(9):4151- 4160. DOI: 10.1210/jcem.86.9.7825
- Singh S, Ilyayeva S. Androgen Insensitivity Syndrome. Treasure Island. StatPearls Publishing; 2021.
- Hughes IA. Consensus statement on management of intersex disorders. Archives of Disease in Childhood. 2005;91(7):554 - 563. DOI: 10.1136/adc.2006.098319
- Lee PA, Houk CP, Ahmed SF, Hughes IA, in collaboration with the participants in the International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus Statement on Management of Intersex Disorders. Pediatrics. 2006;118(2):e488-e500. DOI: 10.1542/peds.2006-0738
- Tyutyusheva N, Mancini I, Baroncelli GI, D’Elios S, Peroni D, Meriggiola MC, et al. Complete androgen insensitivity syndrome: from bench to bed. IJMS. 2021;22(3). DOI: 10.3390/ijms22031264
- Listyasari NA, Robevska G, Santosa A, Bouty A, Juniarto A, Van den Bergen J, et al. Genetic analysis reveals complete androgen insensitivity syndrome in female children surgically treated for inguinal hernia. Journal of Investigative Surgery. 2021;34(2):227-233. DOI: 10.1080/08941939.2019.1602690
- Gulía C, Baldassarra S, Zangari A, Briganti V, Gigli S, Gaffi M, et al. Androgen insensitivity syndrome. ERMPS. 2018;22(12):3873 -3887. DOI: 10.26355/eurrev_201806_15272
- Arora S, Sharma N, Rathi A, Singh K, Sehrawat K. Complete androgen insensitivity syndrome with intra-abdominal seminoma in a phenotypic female: A rare presentation. J Can Res Ther. 2021;17(1):272 - 275. DOI: 10.4103/jcrt.JCRT_302_18
- Batista RL, Costa EMF, Rodrigues A de S, Gomes NL, Faria JA, Nishi MY, et al. Androgen insensitivity syndrome: a review. Archives of Endocrinology and Metabolism. 2018;62(2):227-235. DOI: 10.20945/2359-3997000000031
- Farah S, El Masri D, Hirbli K. Complete androgen insensitivity syndrome in a 13-year-old lebanese child, reared as female, with bilateral inguinal hernia: a case report. J Med Case Reports. 2021;15(1). DOI: 10.1186/s13256-021-02738-0
- Costagliola G, Cosci o di Coscio M, Masini B, Baldinotti F, Caligo MA, Tyutyusheva N, et al. Disorders of sexual development with XY karyotype and female phenotype: clinical findings and genetic background in a cohort from a single centre. J Endocrinol Invest. 2021;44(1):145 -151. DOI: 10.1007/s40618-020-01284-8
- Schwartz GB, Kubo H, Laronda MM. Generation of two human induced pluripotent stem cell lines from a patient with complete androgen insensitivity syndrome with a hemizygous single nucleotide variant in the androgen receptor (AR) gene. Stem Cell Research. 2021;55. DOI: 10.1016/j.scr.2021.102441
- Lanciotti L, Cofini M, Leonardi A, Bertozzi M, Penta L, Esposito S. Different clinical presentations and management in complete androgen insensitivity syndrome (CAIS). IJERPH. 2019;16(7). DOI: 10.3390/ijerph16071268
- Cools M, Looijenga L. Update on the pathophysiology and risk factors for the development of malignant testicular germ cell tumors in complete androgen insensitivity syndrome. Sex Dev. 2017;11(4):175 -181. DOI: 10.1159/000477921
- Coelho ML, Soares E, Freixo M, Brandão P, Marinho C, Rocha J, et al. Complete androgen insensitivity syndrome: a rare case of prenatal diagnosis. Rev Bras Ginecol Obstet. 2021;43(09):710 -712. DOI: 10.1055/s-0041-1735986
- Döhnert U, Wünsch L, Hiort O. Gonadectomy in complete androgen insensitivity syndrome: why and when? Sex Dev. 2017;11(4):171-174. DOI: 10.1159/000478082
- Liu Q, Yin X, Li P. Clinical, hormonal and genetic characteristics of androgen insensitivity syndrome in 39 Chinese patients. Reprod Biol Endocrinol. 2020;18(1). DOI: 10.1186/s12958-020-00593-0
- Xu Z, Peng C, Zhang L, Huang Q. Evaluating complete androgen insensitivity syndrome with a multimodal sonography system. Quant Imaging Med Surg. 2022;12(3):2165-2169. DOI: 10.21037/qims-21-893
- Mattila AK, Fagerholm R, Santtila P, Miettinen PJ, Taskinen S. Gender identity and gender role orientation in female assigned patients with disorders of sex development. Journal of Urology. 2012;188(5):1930 -1934. DOI: 10.1016/j.juro.2012.07.018
Citación recomendada: Contreras Cruz RA, Cortez Salvador LE, Villalta Morales L, Fuentes EJ. Síndrome de insensibilidad completa a andrógenos. Alerta. 2022;5(2):112-117. DOI: 10.5377/alerta.v5i2.14397







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}