
Resumen
Introducción. En este trabajo se describen las primeras secuencias completas del genoma de SARS-CoV-2 a partir de muestras de pacientes salvadoreños. Objetivo. Reportar las primeras secuencias completas del genoma del SARS-CoV-2 de casos procedentes de El Salvador. Metodología. Se realizó una secuenciación masiva en la plataforma Illumina a partir de muestras de secreción nasofaríngea. Resultados. El análisis filogenético determinó que estas muestras pertenecen al clado 20C secundario de 20A que tiene en común la variante de la mutación D614G de la glicoproteína espícula. La mutación S: D614G fue encontrada en las seis secuencias de SARS-CoV-2. En la plataforma GISAID, las secuencias mostraron pertenecer al clado GH linaje pangolín B.1.2 y B.1.370; ambos linajes están presentes en Estados Unidos. Conclusión. Este es el primer reporte de secuencias completas del genoma de SARS-CoV-2 que identifica una variante predominante en Centro y Norteamérica.
Introduction. This work describes the first complete SARS-CoV-2 genome sequencies from samples of Salvadorean patients. Objective. To report the first six complete SARS-CoV-2 genome sequencies of six indigenous cases of COVID-19 detected in El Salvador, from nasopharyngeal discharge samples. Methodology. An NGS next generation sequencing was performed from indigenous samples in the Illumina platform. Results. The phylogenetic analysis showed that these isolated belong to the clade 20C secondary of 20A, which have in common the S: D614G variant; the D614G mutation of the spike glycoprotein was found in the six sequencies of the SARS-CoV-2 genome. Using the GISAID platform, the sequencies were found to belong to the clade GH lineage pangolin B.1.2 y B.1.370, both present in the United States. Conclusion. This is the first report of complete SARS-CoV-2 genome sequencies in El Salvador, and it identifies the predominant variant in Central and North America.
Introducción
El SARS-CoV-2 fue descrito como causante de COVID-19 por primera vez en Wuhan, Hubei, China, el 2 de enero del 2020. En los Estados Unidos de América (EUA), se reportó el primer caso el 20 de enero de 2020, en el estado de Washington D. C., confirmado por los Centros para el Control y la Prevención de Enfermedades de Atlanta (CDC, por sus siglas en inglés)1,2. El 18 de marzo se reporta el primer caso confirmado de infección por SARS- CoV-2 en El Salvador. A nivel mundial, las tasas de nuevos casos y muertes por COVID-19 continuaron aumentando, con casi 4 millones de nuevos casos y 60 000 nuevas muertes registradas. Al 15 de noviembre de 2020, se había notificado a la OMS 53,7 millones de casos confirmados y 1,3 millones de muertes3.
El 3 de febrero se publicó la secuenciación del ARN de una muestra de líquido de lavado broncoalveolar de un paciente que trabajaba en un mercado de Wuhan y que ingresó en el Hospital Central de Wuhan el 26 de diciembre de 2019, mientras experimentaba un síndrome respiratorio severo que incluía fiebre, mareos y tos. Así, se identificó una nueva cepa de virus ARN de la familia Coronaviridae, que se designó en ese momento como coronavirus «WH-Human 1» (y también se le conoció como «2019nCoV»). El análisis filogenético del genoma viral completo (29 903 nucleótidos) reveló que el virus estaba estrechamente relacionado en un 89,1% con un grupo de coronavirus similares al SARS del género Betacoronavirus, subgénero Sarbecovirus, encontrado previamente en murciélagos en China4.
A partir de ese momento varios países comenzaron a publicar las secuencias obtenidas de pacientes locales y al momento de redacción del presente reporte, la última actualización del 27 de noviembre de 2020 publicada en la plataforma Global Initiative on Sharing All Influenza Data (GISAID, por sus siglas en inglés), registraba 3407 genomas, muestreados entre diciembre de 2019 y noviembre de 2020.
Como país es importante poder obtener las secuencias de SARS-CoV-2 a partir de muestras respiratorias de pacientes locales con COVID-19, pues esta información ayudará a comprender en parte el variado espectro clínico y curso de la enfermedad. Aademás, desde el punto de vista de la epidemiología molecular, permite comparar las secuencias entre diferentes poblaciones del mundo con las secuencias del país y analizar la evolución del virus a medida avanza la pandemia. Esto será de mucha utilidad para predecir qué podría pasar en los próximos años en el país y cómo responder ante esa eventualidad. Al principio de la pandemia las secuencias estudiadas han permitido determinar linajes, cadenas de transmisión y rutas de infección.
Estas secuencias completas también nos permiten evaluar la variabilidad genética del virus a nivel local, su evolución y el poder predecir algunos factores pronósticos de los pacientes. De igual forma, aunque algunos estudios estadísticos proponen que las mutaciones descritas en la mayoría de secuencias reportadas hasta el momento no demuestran mayor relación con la virulencia, patogenicidad e incluso transmisibilidad e infectividad viral5, estudios recientes de prestigiosos grupos investigadores han demostrado el aumento del potencial de transmisión de las variantes con la mutación D614G, que predomina a nivel mundial6-9. Los cambios en la secuencia son importantes; sin embargo, las secuencias también permiten evaluar el grado de conservación de las regiones del genoma viral, las cuales son incidentes para el estudio de métodos diagnósticos, blancos de fármacos antivirales, así como en la elaboración y seguimiento de eficacia de las vacunas.
Se reportan las primeras seis secuencias completas del genoma del SARS-CoV-2 de seis casos de COVID-19 detectados en El Salvador, a partir de muestras de secreción nasofaríngea.
Con la descripción de estas primeras secuencias se inicia una serie de publicaciones sobre la vigilancia genómica del virus SARS-CoV-2 que serán útiles para conocer la evolución del virus, desarrollo de terapias y vacunas.
Metodología
Muestras
Se analizaron 50 eluidos de ARN de secreción nasofaríngea obtenida de pacientes positivos a la prueba de qRT-PCR para SARSCoV-2, diagnosticados en el Laboratorio Nacional de Salud Pública del Instituto Nacional de Salud (INS) y la Sección de Virología y Microbiología Molecular, Departamento de Microbiología de la Facultad de Medicina, Universidad de El Salvador. La extracción se realizó con el kit PureLink Viral RNA/DNA Mini Kit Invitrogen de ThermoFisher Scientific. Todos los eluidos fueron conservadas a -80 °C posterior a la extracción.
Se realizó la confirmación diagnóstica por qRT-PCR de los genes E y RdRp con dos protocolos: Charité, Berli/EVAg10 con la AgPath-ID One-Step RT-PCR de ThermoFisher Scientific y con el PowerChek 2019-nCoV Real-time PCR kit (https://www.fda.gov/media/140069/download). Las amplificaciones se realizaron en el QuantStudio 5 Real-Time PCR instrument de Applied Biosystems.
De 50 eluidos se escogieron 6 eludidos con CT inferior a 25 con una concentración ≥50 ng/µL de ARN determinado por fluorometría con Quantus fluorometer, Promega.
Generación de libreríaSe utilizó el método de secuenciación con enfoque de amplicones aplicando una variante de reacciones de PCR multiplex de dos grupos con el kit de Paragon CleanPlex® SARS-CoV-2 by SOPHiA, que ha presentado buenos resultados en estudios comparativos de secuenciación masiva (NGS NextGeneration Sequencing)11,12, siguiendo el protocolo del fabricante. Para la generación de los amplicones se utilizó el termociclador eppendorf Flexlid Mastercycler Nexus gradient.
Secuenciación masiva (NGS)Se utilizó la plataforma MiniSeq Illumina con el MiniSeq Rapid High Output Reagent Kit; como control de calidad interno para la generación, secuenciación y alineación de conglomerados se utilizó PhiX control v3 de Illumina.
Análisis bioinformáticoPara el almacenamiento, análisis y gestión de datos en formato FASQ producto de la secuenciación, se utilizó la plataforma informática Base Space Sequence de Illumina. Para el alineamiento preliminar, determinación de las variantes por mutaciones y asignación preliminar de clados, se utilizó la plataforma bioinformática SOPHiA-DDMV5.7.10. Se realizó alineación de las secuencias obtenidas con secuencias consenso NC_045512 en la plataforma en línea FASTA (ebi.ac.uk/tools/sss/fasta/), para generar archivos compatibles para la generación de propuestas filogenéticas como MEGA-X v10.2. Las seis secuencias obtenidas se cargaron para su análisis de variantes por clado a Nexclade beta v0.8.1; finalmente los resultados de las secuencias fueron enviados a GISAID13 para la determinación de clado y linaje filogenético, ya que esto permitió clasificar y validar los virus secuenciados en una plataforma ya ampliamente reconocida por la comunidad científica global14.
Consideraciones éticas
El protocolo de investigación fue sometido y aprobado por el comité local de ética del Instituto Nacional de Salud; los investigadores no tuvieron acceso a la identidad de los pacientes u otros datos sensibles. Las muestras fueron manejadas por un número de referencia asignado por el Laboratorio Nacional de Salud Pública. Los datos de la secuenciación están disponibles en la plataforma GISAID bajo las siguientes: EPI_ISL_671974, EPI_ISL_671978, EPI_ISL_672012, EPI_ISL_672570, EPI_ ISL_672572, EPI_ISL_672573.
Resultados
En las seis secuencias mediante el alineamiento con el genoma de referencia NC_045512 se observó una concordancia de casi el 100%, confirmándose que las mismas eran positivas a SARS-CoV-2. De igual forma, las seis secuencias dieron positivo a SARS-CoV-2 en el análisis preliminar en la plataforma bioinformática SOPHiA-DDMV5.7.10. Además, se analizaron los genes E, M, N, ORF10, ORF1a, ORF1ab, ORF3a, ORF6, ORF7a, ORF7b, ORF8, S, dentro de los cuales se identificó un total de 27 polimorfismos de un solo nucleótido (SNP), desde 10 y hasta 15 SNP en cada una de las seis secuencias. Algunos de los SNPs comunes entre las secuencias, además la mayoría de los SNP identificados, fueron sin sentido (missense), implicando un cambio de aminoácido. Las secuencias basadas en los SNP identificados se agruparon en un solo patrón variante, impulsado principalmente por SNP ubicados en la región S, NSP2, NS3 y gen S, presentando las mutaciones A23403G (SD614G), C1059T (NSP2T265I) y G25563T (NS3Q57H). En el alineamiento con patrón de mutaciones las seis secuencias fueron clasificadas en el clado Nextstrain 20C (Figura 1-A).
Además, se utilizó GISAID para la filogenia de máxima probabilidad de secuencias muestreadas globalmente de SARSCoV214,15. El resultado de la comparación del patrón de mutaciones comparada con la base de secuencias y de manera particular con la secuencia de referencia hCoV19/Wuhan/WIV04/2019, las seis secuencias fueron clasificadas como clado GISAID GH, linaje pangolín B.1.2 y B.1.370, ambos linajes de EUA (Figura 1-B) coincidente con el 20C de Nextstrain.
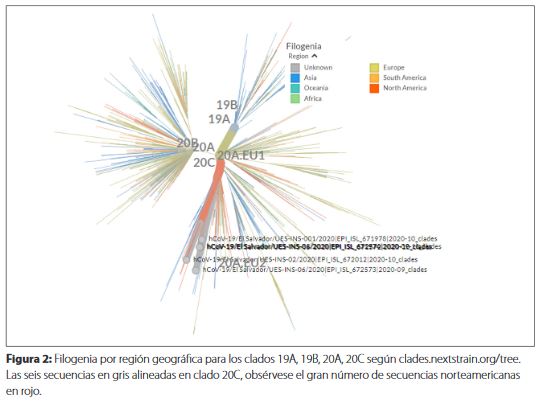
Al rastrear geográficamente el virus por Nextstrain, las seis secuencias mostraron tener mayor similitud con el clado GH (20C) presente en la región norteamericana (Figura 2).
Discusión
Las seis secuencias fueron aceptadas y dadas de alta en GISAID. Esta información de SARS-CoV-2 apoya la calidad del proceso de secuenciación, robustece la vigilancia epidemiológica y permitirá seleccionar para el país la vacuna más representativa.
Con el análisis en la plataforma SOPHiA, ninguna de las mutaciones se asoció a mayor virulencia o patogenicidad; sin embargo, sí se encontró la mutación Spike_D614G que actualmente está siendo ampliamente estudiada por su posible relación con mayor infectividad de la proteína espícula, lo cual aumentaría su transmisibilidad 6.
En el alineamiento con patrón de mutaciones, las seis secuencias fueron clasificadas en el clado Nextstrain 20C, caracterizado por la mutación A23403G, C1059T y G25563T. Esta alcanzó una frecuencia global en torno al 20% en abril, principalmente por secuencias de EUA19; 20C es un clado que se derivó del clado 20A muy frecuente en EUA y que sembró grandes brotes en Europa a principios de 2020, dominando en los posteriores brotes en las Américas. Aademás, una de las características de este clado es la presencia de la mutación D614G, la cual se ha relacionado con aumento de la infectividad viral 9,7 16–18.
Los resultados demostraron que las seis secuencias fueron clasificadas como clado GISAID GH, linaje pangolín B.1.2 y B.1.370, ambos linajes con una alta prevalencia en EUA16–18. Es probable que la migración influya en este patrón, para lo que se requiere obtener más secuencias de todo el territorio nacional, caracterizar epidemiológicamente para conocer su probable origen, documentando la vigilancia molecular del virus y comprender mejor su evolución en el territorio salvadoreño.
Las mutaciones encontradas serán estudiadas a profundidad y se informarán en una próxima entrega. Los SNPs son candidatos fenotípica o epidemiológicamente interesantes para futuras investigaciones, promoviendo combinar con pruebas experimentales la verificación de cualquier fenotipo predicho, como es el caso de la mutación D614G.
Conclusión
Este es el primer reporte de secuencias completas del genoma de SARS-CoV-2 en El Salvador e identifica una variante predominante en Centro y Norteamérica, específicamente el clado GH, linaje pangolín B.1.2 y B.1.370, ambos linajes con una alta prevalencia en EUA. El análisis filogenético evidenció que estas seis muestras pertenecen al clado 20C, clado secundario de 20A, que tiene en común la variante de la mutación D614G de la glicoproteína espícula. Conflicto de interés Los autores declaran no tener conflictos de interés en el estudio ni haber recibido patrocinio de instituciones privadas; la fuente de financiamiento utilizada fueron recursos estatales designados para investigación.
Agradecimientos
Se reconoce el apoyo de las máximas autoridades de la Universidad de El Salvador, Instituto Nacional de Salud y Ministerio de Salud; agradecimientos especiales a la Sección de Virología del Departamento de Microbiología, Facultad de Medicina, Universidad de El Salvador y a la Sección de Virología del Laboratorio Nacional de Salud Pública de El Salvador.
- Harcourt J, Tamin A, Lu X, et al. Severe acute respiratory syndrome coronavirus 2 from patient with coronavirus disease, United States. Emerging Infectious Diseases. 2020;26(6):1266-1273. DOI: 10.3201/ EID2606.200516
- Holshue ML, DeBolt C, Lindquist S, Lofv K.H, Wiesman J, Bruce H, et al. First Case of 2019 Novel Coronavirus in the United States. New England Journal of Medicine. 2020;382(10):929-936. DOI: 10.1056/ nejmoa2001191
- World Health Organization. Weekly Epidemiological Update on COVID-19. World Health Organization. 2020;(3 November):1;4. Disponible en: https://www.who.int/publications/m/item/weekly-epidemiological-update—3-november-2020
- Wu F, Zhao S, Yu B, Chen Y.M, Wang W, Song Z.G, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-269. DOI: 10.1038/s41586-020-2008-3
- Van Dorp L, Richard D, Tan CC, Shaw L, Acman M, Balloux F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nature Communications. Published online 2020. DOI: 10.1101/2020.05.21.108506
- Jackson CB, Zhang L, Farzan M, Choe H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochemical and Biophysical Research Communications. 2020;(xxxx). DOI: 10.1016/j.bbrc.2020.11.026
- Korber B, Fischer WM, Gnanakaran S, Better K, Yoon H, Theiler J, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182(4):812-827.e19. DOI: 10.1016/j.cell.2020.06.043
- Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quilan B.D, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nature Communications. (2020):1-9. DOI:10.1038/ s41467-020-19808-4
- Zhang L, Jackson C, Mou H, Ojha M, Rangarajan E.S, Izard T, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv : the preprint server for biology. Published online 2020. DOI: 10.1101/2020.06.12.148726
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu D.K.W, et al. Detection of 2019 -nCoV by RT-PCR. Euro Surveill. 2020;25(3):1-8. Disponible en: https://www.who.int/docs/default-source/coronaviruse/wuhan-virus-assay-v1991527e5122341d99287a1b17c111902.pdf?sfvrsn=d381fc88_21
- Klempt P, Brož P, Kašný M, Novotný A, Kvapilová K, Kvapil P. Performance of targeted library preparation solutions for SARS-CoV-2 whole genome analysis. Diagnostics. 2020;10(10):1-12. DOI: 10.3390/ diagnostics10100769
- Charre C, Ginevra C, Sabatier M, Regue H, Destras G, Brun S, et al. Evaluation of NGSbased approaches for SARS-CoV-2 whole genome characterisation. Virus Evolution. Published online 2020. DOI:10.1093/ve/ veaa075
- GISAID – phylodynamics. Accessed December 22, 2020. Disponible en: isaid.org/epiflu-applications/phylodynamics/
- Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges. 2017;1(1):33-46. DOI: 10.1002/ gch2.1018
- Rambaut A, Holmes EC, O’Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nature Microbiology. 2020;5(11):1403-1407. DOI: 10.1038/s41564020-0770-5
- Toyoshima Y, Nemoto K, Matsumoto S, Nakamura Y, Kiyotani K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. Journal of Human Genetics. 2020;65(12):1075-1082. DOI: 10.1038/s10038-020-0808-9
- Hodcroft EB, Zuber M, Nadeau S, Candelas F, Standler T, Neher R.A, et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv. 2020;2020(October):2020.10.25.20219063. DOI: https://doi.org/10.1101/2020.10.25.20219063
- Pfefferle S, Günther T, Kobbe R, Czech-Sioli M, Indenbirken D, Robitaille A, et al. SARS Coronavirus-2 variant tracing within the first Coronavirus Disease 19 clusters in northern Germany. Clinical Microbiology and Infection. 2020;(xxxx):2-5. DOI: 10.1016/j. cmi.2020.09.034
- Coiras M, Diaz F, Primo E, Bojo C, Pérez- Gómez B, Grupo de análisis científico de coronavirus del Instituto de Salud Carlos III, Riesgo FDE, La EN, Por E. Informe del Grupo de Análisis Científico de Coronavirus del ISCIII (GACC-ISCIII) factores de riesgo en la enfermedad por SARS-CoV-2 (covid-19) *Este informe está realizado con la evidencia científica disponible en la fecha de su elaboración y podrá ser a. 2020;2. Disponible en: https://fundadeps.org/wp-content/uploads/2020/04/TRANSMISIÓN-DEL-VIRUS_1.pdf
Citación recomendada: Rivera NR, Ortega Pérez CA, Sandoval López X, Hernández Ávila CE. Primeras seis secuencias del genoma completo de SARS-CoV-2 por NGS en El Salvador. Alerta. 2021;4(1):61-66. DOI: 10.5377/ alerta.v4i1.10682







{kind=link}
{kind=link}